Митохондриальная энцефаломиопатия с лактат-ацидозом и инсультоподобными эпизодами мелас (melas). Редкие болезни Дебют melas синдром во взрослом возрасте наблюдение
Ключевые слова
СИНДРОМ MELAS / MELAS SYNDROME / ЭПИЛЕПСИЯ / EPILEPSY / КЛИНИКА / CLINICAL PICTURE / ДИАГНОСТИКА / DIAGNOSTICS / ЛЕЧЕНИЕ / TREATMENTАннотация научной статьи по клинической медицине, автор научной работы - Мухин К.Ю., Миронов М.Б., Никифорова Н.В., Михайлова С.В., Чадаев В.А.
Синдром MELAS генетически детерминированное заболевание из группы митохондриальных болезней, определяется как митохондриальная энцефаломиопатия с лактат-ацидозом и инсультоподобными эпизодами (mitochondrial encephalomyopathy, lactic acidosis with stroke-like episodes). В патологический процесс вовлекаются все органы и ткани, но в большей степени страдают мышечная и нервная системы. Заболевание наиболее часто развивается в возрасте от 6 до 10 лет. Течение болезни прогрессирующее. В большинстве случаев заболевание манифестирует с эпилептических приступов, рецидивирующих головных болей, рвоты, анорексии. Эпилепсия важное клиническое проявление синдрома MELAS. Эпилептические приступы первый распознаваемый симптом при митохондриальных энцефалопатиях (МЭ) в 53% случаев. При MELAS наиболее часто встречается затылочная эпилепсия . С прогрессированием заболевания отмечается резистентность эпилепсии к терапии, нередко со статусным течением. Описаны случаи трансформации в кожевниковскую эпилепсию . Приводим историю болезни пациента с верифицированным при жизни диагнозом синдрома MELAS.
Похожие темы научных работ по клинической медицине, автор научной работы - Мухин К.Ю., Миронов М.Б., Никифорова Н.В., Михайлова С.В., Чадаев В.А.
-
Митохондриальная энцефалопатия с инсультоподобными эпизодами и лактат-ацидозом (синдром melas): критерии диагностики, особенности эпилептических приступов и подходы к лечению на примере клинического случая
2017 / Ямин М.А., Черникова И.В., Арасланова Л.В., Шевкун П.А. -
Инсульты при митохондриальных заболеваниях
2012 / Пизова Н. В. -
Эпилепсия у детей с митохондриальными заболеваниями: особенности диагностики и лечения
2012 / Заваденко Н. Н., Холин А. А. -
Неврологические нарушения при митохондриальной энцефаломиопатии - лактат-ацидозе с инсультоподобными эпизодами (синдроме MELAS)
2012 / Харламов Дмитрий Алексеевич, Крапивкин Алексей Игоревич, Сухоруков Владимир Сергеевич, Куфтина Людмила Андреевна, Грознова Ольга Сергеевна -
Melas-синдром как необычная причина гипопаратиреоза: клиническое наблюдение
2018 / Умярова Диляра Шамилевна, Гребенникова Татьяна Алексеевна, Зенкова Татьяна Станиславовна, Соркина Екатерина Леонидовна, Белая Жанна Евгеньевна -
Инсультоподобные эпизоды при митохондриальной энцефаломиопатии с лактат-ацидозом
2010 / Калашникова Людмила Андреевна, Добрынина Л. А., Сахарова А. В., Чайковская Р. П., Мир-касимов М. Ф., Коновалов Р. Н., Шабалина А. А., Костырева М. В., Гнездицкий В. В., Процкий С. В. -
Митохондриальные цитопатии: синдромы melas и MIDD. Один генетический дефект -разные клинические фенотипы
2017 / Muranova A.V., Strokov I.A. -
Доброкачественная затылочная эпилепсия детского возраста с ранним дебютом(синдром Панайотопулоса). Описание клинического случая
2015 / Матюк Ю.В., Котов А.С., Борисова М.Н., Пантелеева М.В., Шаталин А.В. -
Полиморфизм клинических проявлений прогрессирующей митохондриальной энцефаломиопатии, ассоциированной с мутацией гена POLG1
2016 / Яблонская М.И., Николаева Е.А., Шаталов П.А., Харабадзе М.Н. -
Диагностическая ценность исследования цитохимической активности ферментов при наследственных митохондриальных болезнях
2017 / Казанцева И.А., Котов С.В., Бородатая Е.В., Сидорова О.П., Котов А.С.
EPILEPSY IN MELAS SYNDROME
MELAS syndrome is a genetically determined disease of the mitochondrial group, defined as mitochondrial encephalomyopathy, lactic acidosis with stroke-like episodes. The pathologic process involves all organs and tissues, but it is mostly adversive for the muscular and nervous systems. The disease is most frequent in children aged 6 to 10. The clinical course is progressive. In most cases the disease is manifested by epileptic seizures, relapsing headaches, vomiting, anorexia. The important clinical presentation of MELAS syndrome is epilepsy . Epileptic seizures is the initial diagnosed symptom of mitochondrial encephalopathies (ME) in 53% of cases. Occipital epilepsy is most frequent in MELAS syndrome . As the disease progresses, resistance of epilepsy to treatment is observed, often with occurrence of status epilepticus. Some cases of transformation into Kozhevnikov"s epilepsy are described. A history of a patient with a verified while alive diagnosis of MELAS syndrome is given.
Текст научной работы на тему «Эпилепсия при синдроме melas»
ТОМ IV ВЫПУСК 3 2009
ЭПИЛЕПСИЯ ПРИ СИНДРОМЕ MELAS
К.Ю. Мухин1, М.Б. Миронов1, Н.В. Никифорова1, C.B. Михайлова2, ВА. Чадаев1, АА. Алиханов1-2, Б.Н. Рыжков1, А.С. Петрухин1
EPILEPSY IN MELAS SYNDROME
KYu. Mukhin1, M.B. Mironov1, N.V. Nikiforova1, S.V. Mikhailova2, УА. Chadaev1, АА. Alikhanov1-2, B.N. Ryzkov1, AS. Petrukhin1
1 - Кафедра неврологии и нейрохирургии педиатрического факультета ГОУ ВПО РГМУ Росздрава
2 - Российская детская клиническая больница
Синдром MELAS - генетически детерминированное заболевание из группымитохондриалъных болезней, определяется какмитохондриалъная энцефаломиопатия слактат-ацидозом и инсулътоподобными эпизодами (mitochondrial encephalomyopathy, lactic acidosis with stroke-like episodes). В патологический процесс вовлекаются все органы и ткани, но в болъшей степени страдают мышечная и нервная системы. Заболевание наиболее часто развивается в возрасте от 6 до 10 лет. Течение болезни прогрессирующее. В болъшинстве случаев заболевание манифестирует с эпилептических приступов, рецидивирующих головных болей,рвоты, анорексии. Эпилепсия - важное клиническое проявление синдрома MELAs. Эпилептические приступы - первый распознаваемый симптом при митохондриалъных энцефалопатиях (МЭ) в 53% случаев. При MELAS наиболее часто встречается затылочная эпилепсия. С прогрессированием заболевания отмечается резистентностъ эпилепсии к терапии, нередко со статусным течением. Описаны случаи трансформации в кожевниковскую эпилепсию. Приводим историю болезни пациента с верифицированным при жизни диагнозом синдрома MELAS.
Ключевые слова: синдром MELAS, эпилепсия, клиника, диагностика, лечение.
MELAS syndrome is a genetically determined disease of the mitochondrial group, defined as mitochondrial encephalomyopathy, lactic acidosis with stroke-like episodes. The pathologic process involves all organs and tissues, but it is mostly adversive for the muscular and nervous systems. The disease is most frequent in children aged 6 to 10. The clinical course is progressive. In most cases the disease is manifested by epileptic seizures, relapsing headaches, vomiting, anorexia. The important clinical presentation of MELAS syndrome is epilepsy. Epileptic seizures is the initial diagnosed symptom of mitochondrial encephalopathies (ME) in 53% of cases. Occipital epilepsy is mostfrequent in MELAS syndrome. As the disease progresses, resistance of epilepsy to treatment is observed, often with occurrence of status epilepticus. Some cases of transformation into Kozhevnikov"s epilepsy are described. A history of a patient with a verified while alive diagnosis of MELAS syndrome is given.
Key words: MELAS syndrome, epilepsy, clinical picture, diagnostics, treatment.
Синдром MELAS - генетически детерминированное заболевание из группы митохондриальных болезней, определяется как митохондриальная энцефаломиопатия с лактат-ацидозом и ин-сультоподобными эпизодами (mitochondrial encephalomyopathy, lactic acidosis with stroke-like episodes).
Синдром MELAS впервые был выделен в самостоятельную нозологическую форму S. Pavlakis и соавт. в 1984 году . Однако ряд авторов предполагает, что заболевание было описано раньше под названием «семейная полиодистрофия, митохондриальная миопатия, лактацидемия».
Распространенность в популяции не установлена. К 2000 г. опубликовано более 120 наблюдений синдрома MELAS, в том числе и в отечественной печати .
Синдром MELAS в 25% случаев наследуется по материнской линии с высоким риском, однако у 56-75% больных семейный анамнез не отягощен. Заболевание связано с мутациями генов митохондри-альной ДНК, кодирующих субъединицы комплексов дыхательной цепи и гены транспортных РНК (MT-ND1, MT-ND5, MT-TH, MT-TL1 и MT-TV). В 80-90% случаев синдрома MELAS в основе заболевания лежит точковая мутация в гене MT-TL1, кодирующем транспортную РНК лейцина . При данной мутации происходит замена нуклеотида аденина на гуанин в позиции 3243 (A3243G), что нарушает синтез всех белков в митохондриях .
В патологический процесс вовлекаются все органы и ткани, но в большей степени страдают мышечная и нервная сис-
Мухин К.Ю., Миронов М.Б., Никифорова Н.В., Михайлова C.B., Чадаев ВА., Алиханов АА., Рыжков БН., Петрухин А.С.
Эпилепсия при синдроме MELAS Рус. жур. дет. невр.: т. IV, вып. 3, 2009.
ОРИГИНАЛЬНЫЕ СТАТЬИ
темы, как наиболее энергозависимые. Тяжесть клинических проявлений зависит от порогового эффекта (возраста, потребности тканей в энергии), от контроля ядерных генов за синтезом дыхательной цепи, гетероплазмии (разного содержания в тканях мутантных молекул мтДНК). Показано, что у больных синдромом MELAS содержание мутантной мтДНК в различных тканях составляет 93-96%. У членов семьи пробандов в тканях также определяется мутантная мтДНК, однако ее содержание существенно ниже: 62-89% при стертой форме болезни, от 28 до 89% - при отсутствии клинических признаков синдрома.
Заболевание наиболее часто развивается в возрасте от 6 до 10 лет, но встречаются случаи более раннего (до двух лет) или более позднего дебюта - от 21 до 40 лет. До начала заболевания 90-100% пациентов развиваются нормально. Течение болезни прогрессирующее, более злокачественное при раннем дебюте .
В большинстве случаев заболевание манифестирует с эпилептических приступов, рецидивирующих головных болей, рвоты, анорексии. Также следует обращать внимание на непереносимость физических нагрузок в виде ухудшения самочувствия и появления мышечной слабости. Миопатический симптомо-комплекс проявляется непереносимостью физических нагрузок, мышечной слабостью, утомляемостью, иногда - ги-потрофиями мышц .
По мере прогрессирования болезни обычно развивается деменция. Такие симптомы, как мозжечковая атаксия, ней-росенсорная глухота, периферическая полиневропатия встречаются реже.
Характерны инсультоподобные эпизоды, которые могут проявляться рецидивирующими приступами головной боли, головокружением, развитием очаговой неврологической симптоматики (парезы, гемианопсия), коматозными состояниями. Подобные острые эпизоды часто провоцируются лихорадкой или интер-куррентными инфекциями. Эти проявления могут иметь довольно быстрый регресс (от нескольких часов до нескольких недель), а также склонность к рецидиви-рованию .
Эпилепсия является важным клиническим проявлением, часто возникающим на ранних этапах синдрома MELAS. Это
нередко наиболее явное неврологическое проявление, особенно при нетипичных митохондриальных энцефалопати-ях (МЭ) . Эпилептические приступы - первый распознаваемый симптом при митохондриальных энцефалопатиях (МЭ) в 53% случаев .
При MELAS наиболее часто встречается затылочная эпилепсия (ЗЭ). Характерны фокальные приступы, исходящие из затылочных долей . Приступы нередко ассоциируются с транзиторными или постоянными неврологическими симптомами, такими как выпадение полей зрения .
Приступы, исходящие из затылочной коры, подразделяются по своим проявлениям на субъективные ощущения (аура) и на клинически выявляемые симптомы, как правило, с моторным компонентом. К эпилептическим аурам, исходящим из затылочной доли, относят простые и сложные зрительные галлюцинации, амавроз. Наиболее типичными приступами, характерными для ЗЭ, являются простые зрительные галлюцинации, которые могут проявляться позитивными (вспышки, пятна света) и негативными симптомами (скотома, гемианопсия). Наиболее часто зрительные галлюцинации описываются как пятно или пятна света, постоянные или мигающие. Как правило, пятно имеет белый цвет с зеленоватым оттенком . Также галлюцинации могут иметь разноцветный или монохроматический характер. Галлюцинации обычно появляются в полях зрения контралатерально очагу возбуждения в затылочной коре с последующим распространением. Однако, следует отметить, что в жалобах пациентов зрительная аура выявляется не часто.
Сложные зрительные галлюцинации отмечаются при распространении эпилептического возбуждения на окципито-темпоральные или окципито-париеталь-ные отделы. Сложные зрительные галлюцинации могут проявляться в форме людей, животных объектов или сцен, быть хорошо знакомыми или незнакомыми, приятными или устрашающими, отпугивающими, простыми или гротескными, могут быть статичными или передвигаться в горизонтальной плоскости и исчезать. Как правило, они являются терминальным симптомом перед развитием моторного приступа; могут быть первым иктальным симптомом, но чаще возникают вслед за
ТОМ IV ВЫПУСК 3 2009
элементарными галлюцинациями.
Особым, крайне тяжелым для диагностики типом приступов, исходящих из затылочной коры, является иктальный ама-вроз. По мнению многих авторов, это такой же частый симптом раздражения затылочной доли, как и зрительные галлюцинации, но нередко остается нераспознанным. Обычно пациенты не выделяют этот симптом отдельно в структуре приступа. Потеря зрения возникает билатерально с выпадением боковых полей. Возможна гомонимная гемианопсия кон-тралатерально очагу приступа. Ощущения пациентов описываются ими как потемнение в глазах, «белая тьма», нарушение восприятия цвета. Возможно статусное течение с формированием так называемого status epilepticus amauroticus.
Затылочные приступы могут проявляться вегетативными симптомами. К ним относят головную боль мигренозно-го характера, головокружение, тошноту, рвоту. Частый симптом - постприступ-ная мигренеподобная головная боль.
Клинические проявления приступов, возникающих ограниченно в затылочной коре, характеризуются девиацией глаз в сторону. Девиация глаз может отмечаться совместно с отклонением головы в сторону. В большинстве случаев отмечается девиация глаз в сторону контра-латеральную очагу. Однако описаны случаи, когда отведение глаз наблюдается в сторону очага. Также одна из особенностей «затылочных» приступов - мгновенное распространение разряда на передние отделы головного мозга, при этом в клинической картине, как правило, преобладает выраженный моторный компонент. Возможны тонические, тонико-кло-нические (как гемиконвульсивные, так и вторично-генерализованные), аутомо-торные приступы . В связи с этим, важно выявлять начальные клинические симптомы - немотивируемая и внезапная остановка взора, разглядывание несуществующих предметов, беспричинная улыбка, вегетативные проявления и обязательно документальное подтверждение первичной иктогенной зоны методом ВЭМ.
С прогрессированием заболевания отмечается резистентность эпилепсии к терапии, нередко со статусным течением. Описаны случаи трансформации в ко-жевниковскую эпилепсию . Ряд авто-
ров описывает возможность возникновения эпилептического статуса как первого симптома у пациентов с MELAS без предшествующих эпилептических приступов в анамнезе . Ribacoba R. и соавт. описывают в своей публикации 4 случая развития epilepsia partialis continua с фокальными моторными приступами, которой предшествовали в анамнезе эпизоды ми-гренозной головной боли. Miyazaki M. и соавт. показали возможность продолженного фокального миоклонуса в рамках epilepsia partialis continua у пациентов с MELAS . Araki T. и соавт. наблюдали пациентку в возрасте 37 лет с эпилептическим статусом фокальных приступов в виде флюктуации сознания, гомонимной гемианопсии в сочетании с пароксиз-мальными эпизодами девиации глаз в сторону . На ЭЭГ регистрировались продолженные ЭЭГ-паттерны приступов, локализованные в затылочной области. У взрослых пациентов с MELAS наблюдается преобладание фокальных моторных приступов, но на ЭЭГ отмечается преобладание мультирегиональной эпилепти-формной активности в затылочных областях .
Эпилептиформная активность регистрируется в 71% случаев после дебюта приступов . При электроэнцефалографическом исследовании пациентов с MELAS синдромом характерна эпилепти-формная активность в затылочных областях . Ряд авторов связывает появление региональных эпилептиформных нарушений с инсультами. По данным исследования Fujimoto S., в остром периоде (т.е. в течение 5 дней после инсультопо-добного эпизода) у большинства обследуемых пациентов с MELAS синдромом отмечались региональные высокоамплитудные дельта-волны в сочетании с полиспайками. Авторы предлагают рассматривать данный паттерн как патогномо-ничный для инсультоподобных эпизодов . Кроме затылочных регионов, эпи-лептиформная активность может распространяться на височные отделы , бифронтально , а также билатерально на задние отделы с диффузным распространением . Возможно появление фотопароксизмального ответа при проведении ритмической фотостимуляции .
Ведущим лабораторным признаком служит повышение уровня лактата в кро-
ОРИГИНАЛЬНЫЕ СТАТЬИ
ви свыше 2,0 ммоль/л, что приводит к развитию лактат-ацидоза.
МРТ головного мозга на ранних стадиях болезни может быть без особенностей, даже при возникновении эпилепсии . Методы нейровизуализации выявляют зоны инфарктов в области больших полушарий (80%), реже - в области мозжечка и базальных ганглиев. Может наблюдаться также кальцификация ба-зальных ганглиев, атрофия коры больших полушарий. При фотонно-эмисси-онном исследовании накопление изотопа обнаруживается за 3-16 дней до появления зоны инфаркта (снижение изотопного сигнала) на компьютерной томограмме головного мозга. МРТ головного мозга демонстрирует области поражения, преимущественно локализованные в затылочных долях, которые могут иметь транзиторный характер . Преимущественно страдает затылочная кора, белое вещество повреждается в меньшей степени. На Т2 взвешенных изображениях очаги поражения мозга при МЕЬАЯ выглядят как зоны повышения интенсивности сигнала . Транзиторные гиперинтенсивные области ряд авторов связывает с обратимым васкулярным отеком .
Ангиография обычно не выявляет сосудистых нарушений . МРТ в режиме диффузно-взвешенного изображения демонстрирует изменения, связанные с вазогенным отеком .
Гистопатология: при исследовании мышечного биоптата выявляются волокна с рваными «красными краями». При аутопсии мозга характерно сочетание старых и новых очагов инфарктов, а также атрофия коры с фокальными очагами некроза.
В настоящее время терапия носит поддерживающий характер. Основное направление лечения - улучшение энергетического баланса митохондрий и дыхательной цепи. Применяют коэнзим р10 (80-300мг/сут), витамины К1 и КЗ (25 мг/сут), янтарную кислоту (до 6 г/сут), витамин С (2-4 г/сут), рибофлавин (100 мг/сут) и никотинамид (до 1 г/сут). В связи с развивающимся вторичным дефицитом карнитина, больным назначается Ь-карнитин (до 100 мг/кг/сут). В качестве антиоксидантной терапии применяют витамин Е (300-500 мг/сут) и витамин С (2-4 мг/сут).
Общепринятых схем антиэпилептической терапии при МЕЬАЯ не существует. Ряд авторов предлагает исключить препараты, способные ингибировать энергетический метаболизм (барбитураты, препараты вальпроевой кислоты; а также и некоторые препараты других групп, например, хлорамфеникол) . В литературе описано несколько отдельных случаев аггравации судорожных приступов при применении вальпроевой кислоты при синдроме МЕЬАЯ с мутацией А3243С . Основными АЭП в лечении эпилепсии при синдроме МЕЬАЯ считаются тег-ретол (или трилептал), топамакс, кеппра в средних терапевтических дозах . Правильно подобранная терапия приводит к существенному урежению частоты вторично-генерализованных судорожных приступов. Однако приступы с нарушением вегетативно-висцеральных и зрительных функций, как правило, резистентны к лечению. В терминальной стадии заболевания частота эпилептических приступов может уменьшаться.
Приводим историю болезни пациента с верифицированным при жизни диагнозом синдрома МЕЬАЯ.
Больной Ч.А., 11 лет, наблюдался в Центре детской неврологии и эпилепсии. При поступлении предъявлялись жалобы на постепенную утрату речевых навыков, выраженное нарушение походки с отказом ходить, значительное снижение зрения, капризность, негативное поведение; ежедневные серийные приступы в виде подергиваний мускулатуры лица, мышц верхних и нижних конечностей, а также кратковременные эпизоды потери зрения.
Дебют, заболевания отмечался в 5 лет 9 мес. Впервые на фоне полного здоровья при засыпании появилась сильная головная боль, простые зрительные галлюцинации («желтый лучик») с последующим насильственным поворотом глаз и головы в сторону и развитием генерализованного тонико-клонического судорожного приступа, после которого отмечалась рвота. Через 9 мес. приступы с такой же симптоматикой повторились и быстро приобрели серийный характер. После назначения тегретола в дозе 400 мг в сутки частота приступов уменьшилась до 1 раза в месяц. Тегретол был заменен на депакин хроно в дозе 900 мг/сут, на фоне которого отмечалась клиническая ремиссия в течение 6 мес. Учитывая клиническую симп-
ТОМ IV ВЫПУСК 3 2009
томатику, приуроченность приступов к периоду засыпания, нормальный интеллект пациента, положительную реакцию на вальпроаты, была диагностирована идиопатическая затылочная эпилепсия.
В 7 лет возобновились фокальные вер-сивные приступы со вторичной генерализацией при засыпании с прежней частотой 1 раз в мес. Повышение дозы депа-кина до 1500 мг/сут не привело к снижению частоты приступов. При добавлении ламиктала в дозе 75 мг/сут приступы купировались на 4 мес., затем возобновились с прежней частотой. В 8 лет присоединились приступы с кратковременной потерей зрения. С 8 лет 8 мес. перед засыпанием стали возникать атипичные аб-сансы: быстрое моргание с прикрыванием век и заведением глазных яблок кверху; сознание флюктуирует.
В 9 лет появились многократные серийные приступы, продолжающиеся в течение нескольких дней, с простыми зрительными галлюцинациями в виде мелькания «лучика» перед глазами, с поворотом глаз и головы вправо. Перед засыпанием подобные приступы иногда переходили в фокальные гемиклонические, которые проявлялись сведением лицевой
мускулатуры справа, подергиванием головы вправо, клониями правых конечностей (больше руки). Иногда после приступа возникала сильная головная боль и рвота. В этом же возрасте появились тормозные приступы: аура в виде ощущения мурашек в большом пальце правой стопы с последующей кратковременной слабостью правой ноги и неловкостью правой руки. В схему лечения введен топамакс в дозе 100 мг/сут - в течение 1 года эпилептических приступов не было.
Также в 9 лет впервые появились приступообразные состояния, сопровождающиеся сильной головной болью, рвотой и развитием правостороннего геми-пареза. В некоторых случаях подобные состояния сопровождались амаврозом продолжительностью от нескольких минут до нескольких дней.
В возрасте 10,5 лет вновь появились приступы в виде поворота головы влево, толчкообразных движений глазных яблок влево, длительностью до 5 с, частотой до 3-х раз в течение часа, ежедневно, даже во время сна. Доза топамакса была увеличена до 150 мг/сут без существенного эффекта. В 10 лет 10 мес. после интенсивной головной боли возникли альтерни-
Рис. 1. Пациент Ч.А. 10 лет. Диагноз: МЕЬАЭ синдром. Симптоматическая фокальная эпилепсия.
Видео-ЭЭГ мониторинг (2004 г.): на фоне диффузного замедления основной активности головного мозга регистрируется продолженная эпилептиформная активность в левой затылочной области. Также зарегистрированы субклинические ЭЭГ-паттерны приступа в левой затылочной области с распространением на левую заднюю височную область.
Центр детской Неврологии и Эпилепсии
поа руководством профессора К.Ю. Мухина занимается диагностикой и печением боаезней нервной системы у аетей, специализируется на аетских формах эпилепсии.
Основные направления
деятельности:
Эпилепсия у детей и подростков
Головная боль
Нарушения сна у детей
Тики, энурез
Обследование детей первых ^ месяцев жизни.
Обследования в нашем центре:
Диагностика и лечение заболеваний нервной системы у детей
Полная диагностика (в том числе прехирургическая) и лечение эпилепсии
Консультация неврологов и эпилептологов
Консультация педиатра (часто болеющие дети, гастроэнтерология и др.)
Консультация психиатра и психолога.
Консультация генетика с проведением анализов (в том числе кариотипирование)
Видео-ЭЭГ-мониторинг (в специально оборудованных палатах Центра или с выездом на дом к пациенту)
Компьютерная (цифровая) электроэнцефалография
УЗДГ (ультразвуковая допплерография) сосудов головы и шеи
Эхоэнцефалография (ЭХО ЭГ)
На нашем сайте вы можете подписаться на журнал «Русский журнал детской неврологии» через Интернет.
Подробная информация о работе Центра с 10:00 до 19:00 по телефонам:
Тел.: (+7495)983-09-03; (+7926)290-50-30 Тел./факс: (+7495) 394-82-52
Адрес: Ул. Борисовские пруды, д. 13, корп. 2. Интернет: www.epileptologist.ru E-mail: [email protected] (подробную схему проезда см. на сайте)
ТОМ IV ВЫПУСК 3 2009
рующие фокальные гемнклоническне и вторично-генерализованные приступы, которые стали серийными и продолжались 48 часов. К топамаксу был добавлен фризиум в дозе 10 мг/сут с временным положительным эффектом.
С 8 лет стали отмечаться сложности с усвоением школьного материала; снизилась память. Появилась повышенная утомляемость, истощаемость, заторможенность мыслительной деятельности. Мальчик стал капризным, раздражительным, негативным; снизился фон настроения. С 9 лет отмечалось усиление данной симптоматики.
Из анамнеза жизни известно, что ребенок родился от второй нормально протекавшей беременности, вторых срочных родов, вес при рождении 2800 г, длина 53 см. Раннее психомоторное и речевое развитие полностью соответствовало возрасту. Перенесенные заболевания: ветряная оспа в 6 лет, частые ОРВИ (до 4 раз в год) с 6 лет. Наследственность по эпилепсии и другим неврологическим заболеваниям не отягощена.
На момент осмотра (11 лет) состояние ребенка тяжелое; на осмотр реагирует негативно. В сознании, ориентирован в про-
странстве и времени. В контакт вступает крайне неохотно, инструкции выполнять отказывается. Спонтанный нистагм влево, голова наклонена к левому плечу с поворотом вправо. Язык по средней линии, глоточный рефлекс снижен; отмечается дисфагия, дизартрия. Зрение снижено.
Определяется умеренная диффузная мышечная гипотония. Сухожильные рефлексы равномерно снижены. Отмечено легкое снижение мышечной силы в правых конечностях. Патологических стопных рефлексов не выявлено. Объективных данных за нарушение чувствительности нет. В пробе Ромберга не стоит. Отказывается ходить. При попытке поставить на ноги - плачет, присаживается на пол. Мимопопадание при выполнении пальце-указательной пробы. Говорит медленно, отдельными словами, с неохотой.
Дополнительные методы обследования. Видео-ЭЭГ мониторинг (2004 год). Значительное замедление основной активности фоновой записи. В ходе исследования регистрируется продолженная эпилептиформная активность в левой затылочной области с распространением на левую задневисочную и с периодическим формированием ЭЭГ паттерна при-
1993 г.р. 16/12/05
Рис. 2. Пациент Ч.А. 11 лет. Диагноз: MELAS синдром. Симптоматическая фокальная эпилепсия.
Видео-ЭЭГ мониторинг проведен в динамике через 1 год (2005 г.): значительное замедление фоновой активности головного мозга. В ходе записи сна регистрируется продолженное региональное замедление в правой лобно-центральной области, в структуре которого выявляется пик-волновая активность в правой лобно-центральной области.
ОРИГИНАЛЬНЫЕ СТАТЬИ
ступа (рис. 1). Также определяется продолженное региональное замедление в правой лобно-центральной области с включением единичных острых волн.
Видео-ЭЭГ мониторинг в динамике (2005 год): Значительное замедление фоновой активности головного мозга. В ходе исследования регистрируется продолженное региональное замедление в правой лобно-центральной области. В структуре регионального замедления в правой лобно-центральной области выявляется пик-волновая активность (рис. 2).
МРТ головного мозга. На первом МРТ (6 лет) был обнаружен единичный гиперинтенсивный сигнал в Т2-режиме в левой гемисфере мозжечка. МРТ исследование в динамике (10,5 лет): выявлено значительное ухудшение первичного поражения с распространением патологического процесса широко на левую и правую заты-лочно-теменные области обеих гемисфер головного мозга (профессор A.A. Алиха-нов).
Зрительные вызванные потенциалы: значительные морфофункциональные изменения в зрительной афферентной системе на уровне зрительного нерва и коркового отдела зрительного анализатора, более выраженные слева.
Консультация офтальмолога: частичная атрофия зрительных нервов. Элементы корковой агнозии.
Электрокардиограмма: ритм эктопический с ускорением до 100 ударов в мин.
Вертикальное положение электрической оси сердца. Изменение процессов репо-ляризации, которые в ортостазе более выражены.
Электронейромиография: выявлен первично-мышечный тип поражения. Скорости проведения по периферическим нервам не снижены.
Исследование уровня лактата в крови: содержание лактата в крови 3,0 ммоль/л (норма - до 1,8).
Учитывая наличие эпилептических приступов, исходящих из затылочных отделов коры большого мозга, резистентных к терапии, инсультоподобных эпизодов, периодов амавроза, снижение когнитивных функций, наличие на МРТ гиперинтенсивных сигналов в мозжечке и задних отделах коры головного мозга, повышение уровня лактата в крови, у пациента был предположен диагноз синдрома MELAS. В ходе генетического обследования в клетках крови обнаружена мутация A3243G в гетероплазмическом состоянии (диагностика проводилась в ГУ МГНЦ РАМН), и диагноз был верифицирован.
Наблюдение в катамнезе показало быстрое прогрессирование нарушений высших психических функций, развитие корковой слепоты, полной обездвиженности пациента с последующим наступлением летального исхода в возрасте 12 лет 10 мес. (спустя 7 лет с момента дебюта заболевания).
Библиография
1. Николаева Е.А., Темин П.А. Митохондриальные болезни, сопровождающиеся нарушением нервно-психического развития. Синдром MELAS // Наследственные нарушения нервно-психического развития детей. Руководство для врачей под редакцией Темина П.А. Казанцевой Л.З. - Медицина, 2001. - С. 96-107.
2. Николаева Е.А., Темин ПА., Никанорова М.Ю., Клембовский А.И., Сухоруков В.С., Дорофеева М.Ю., Кор-сунский А.А. Лечение ребенка с митохондриальным синдромом MELAS (митохондриальная энцефалопатия, лактат-ацидоз, инсультоподобные эпизоды) // Российский вестник перинатологии и педиатрии. - 1997. - №2. - С. 30-34.
3. Смирнова И.Н., Кистенёв Б.А., Кротенкова М.В., Суслина ЗА. Инсультоподобное течение митохондриаль-ной энцефаломиопатии (синдром MELAS) // Атмосфера. Нервные болезни. - 2006. - №1. - С. 43-48.
4. Темин ПА, Никанорова М.Ю., Николаева Е.А. Синдром MELAS (митохондриальная энцефаломиопатия, лактат-ацидоз, инсультоподобные эпизоды): основные проявления, критерии диагностики, возможности лечения // Неврол. журн. - 1998. - №2. - С. 43-48.
5. Ajmone-Marsan C., Ralston B. The epileptic seizure, its functional morphology and diagnostic significance. - Springfield (IL): Charles C. Thomas, 1957. - P. 3-231.
6. Aldrich M.S., Vanderzant C.W., Alessi A.G., Abou-Khalil B., Sackellares J.C. Ictal cortical blindness with permanent visual loss // Epilepsia. - 1989. - V. 30. - P. 116-20.
7. Araki T., Suzuki J., Taniwaki Y., Ishido K., Kamikaseda K., Turuta Y., Yamada T. A case of MELAS presenting complex partial status epilepticus // Rinsho Shinkeigaku. - 2001. - V. 41(8). - P. 487-90.
ТОМ IV ВЫПУСК 3 2009
8. Canafoglia L., Franceschetti S., Antozzi C., Carrara F., Farina L., Granata T., Lamantea E., Savoiardo M., Uziel G., Villani F., Zeviani M., Avanzini G. Epileptic phenotypes associated with mitochondrial disorders // Neurology. - 2001. - V. 56(10). - P. 1340-6.
9. Chih-Ming Lin, Peterus Thajeb. Valproic acid aggravates epilepsy due to MELAS in a patient with an A3243G mutation of mitochondrial DNA // Metab Brain Dis. - 2007 - V. 22(1). - P. 105-109.
10. Chinnery P.F., Howell N., Lightowlers R.N. et al. Molecular pathology of MELAS and MERRF. The relationship between mutation load and clinical phenotypes // Brain. - 1997. - V.120. - P. 1713-1721.
11. Durand-Dubief F., Ryvlin P, Mauguiere F. Polymorphism of epilepsy associated with the A3243G mutation of mitochondrial DNA (MELAS): reasons for delayed diagnosis // Rev Neurol (Paris). - 2004. - V. 160(8-9). - P. 824-829.
12. Dvorkin G., Andermann F., Carpenter S. Classical migraine, intractable epilepsy and multiple strokes: a syndrome related to mitochondrial encephalopathy / In: Andermann F., Lugaresi E., editors. Migraine and epilepsy. - Boston: Butterworths, 1987. - P. 203-32.
13. Fujimoto S., Mizuno K., Shibata H., Kanayama M., Kobayashi M., Sugiyama N., Ban K., Ishikawa T., Itoh T., Togari H., Wada Y. Serial electroencephalographs findings in patients with MELAS // Pediatr Neurol. - 1999. - V. 20(1). - P. 43-48.
14. Goto Y., Nonaka I., Horai S.A. A mutation in the tRNA leu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies // Nature. - 1990. - V. 348. - P. 651-653.
15. Hasuo K., Tamura S., Yasumori K., Uchino A., Goda S., Ishimoto S., et al. Computed tomography and angiogra-phy in MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes): report of 3 cases // Neuroradiology. - 1987. -V. 29. - P. 393-397.
16. Hirano M., Pavlakis S.G. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke like episodes (MELAS): Current concepts // J. clin. Neurol. - 1994. - V. 9. - P. 4-13.
17. Hori A., Yoshioka A., Kataoka S., Furui K., Tsukada K., Kosoegawa H., Sugianto , Hirose G. Epileptic seizures in a patient with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) // Jpn J Psychiatry Neurol. - 1989. - V. 43(3). - P. 536-537.
18. Kuriyama M., Umezaki H., Fukuda Y., Osame M., Koike K., Tateishi J., et al. Mitochondrial encephalomyopathy with lactate-pyruvate elevation and brain infarctions // Neurology. - 1984. - V. 34. - P. 72-77.
19. Kuzniecky R. Symptomatic occipital lobe epilepsy // Epilepsia. - 1998. - V. 39 Suppl 4. - P. 24-31.
20. Ludwig B.I., Ajmone-Marsan C., Van Buren J. Depth and direct cortical recording in seizure disorders of extratemporal origin // Neurology. - 1976. - V. 26. - P. 1085-1099.
21. Ludwig B.I., Ajmone-Marsan C. Clinical ictal patterns in epileptic patients with occipital electroencephalo-graphic foci // Neurology. - 1975. - V. 25. - P. 463-471.
22. Matthews P.M., Tampieri D., Berkovic S.F., Andermann F., Silver K., Chityat D., et al. Magnetic resonance imaging shows specific abnormalities in the MELAS syndrome // Neurology. - 1991. - V. 41. - P. 1043-1046.
23. Miyazaki M., Saijo T., Mori K., Tayama M., Naito E., Hashimoto T., Kuroda Y., Nonaka I. A case with MELAS associated with epilepsia partialis continua // No To Hattatsu. - 1991. - V. 23(1). - P. 65-70.
24. Montagna P., Gallassi R., Medori R., Govoni E., Zeviani M., Di Mauro S., et al. MELAS syndrome: characteristic migrainous and epileptic features and maternal transmission // Neurology. - 1988. - V. 38. - P. 751-754.
25. Ooiwa Y., Uematsu Y., Terada T., Nakai K., Itakura T., Komai N., et al. Cerebral blood flow in mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes // Stroke. - 1993. - V. 24. - P. 304-309.
26. Pavlakis S.G., Phillips P.C., Di Mauro S. et al. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes: A distinctive clinical syndrome // An neurol. - 1984. - V. 16. - P. 481-488.
27. Ribacoba R., Salas-Puig J., Gonzalez C., Astudillo A. Characteristics of status epilepticus in MELAS. Analysis of four cases // Neurologia. - 2006. - V. 21(1). - P. 1-11.
28. Williamson P.D., Spencer S.S. Clinical and EEG features of complex partial seizures of extratemporal origin // Epilepsia. - 1986. - V. 27 (Suppl 2). - P. 46-63.
29. Williamson P.D., Thadani V.M., Darcey T.M., Spencer D.D., Spencer S.S., Mattson R.H. Occipital lobe epilepsy: clinical characteristics, seizure spread patterns, and results of surgery // Ann Neurol. - 1992. - V. 31. - P. 3-13.
30. Yi-Min Chen, Chih-Ming Lin, Peterus Thajeb. Paradoxical effect of sodium valproate that aggravates epilepsy of MELAS in a patient with A3243G mutation of the mitochondrial DNA // Central European Journal of Medicine. - 2007. - V. 2(1). - P.103-107.
31. Yoneda M., Maeda M., Kimura H., Fujii A., Katayama K., Kuriyama M. Vasogenic edema on MELAS: a serial study with diffusion-weighted MR imaging // Neurology. - 1999. - V. 53. - P. 2182-2184.
Синдром МЕЛАС — это митохондриальное заболевание, характеризующееся поражением мышц и ЦНС.
MELAS (англ. Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes - «митохондриальная энцефаломиопатия, лактатацидоз, инсультоподобные эпизоды») - прогрессирующее нейродегенеративное заболевание, характеризующееся проявлениями, перечисленными в названии, и сопровождается полиморфной симптоматикой - инсульта, диабетом, судорогами, снижением слуха, сердечными заболеваниями, низким ростом, эндокринопатиями, непереносимостью физических нагрузок и нейропсихиатрическими отклонениями.
История
.
Синдром MELAS был впервые описан в 1984 году Павлакисом и коллегами; десять лет спустя Павлакис и Мицио Хирано опубликовали обзор 110 случаев заболевания.
Тип наследования:
материнский
Эпидемиология :
Точная частота заболевания не известна. В литературе имеются единичные данные о частоте заболевания. На севере Финляндии частота мутации A3243G, составляет 16.3:100 000.
Патогенез :
Мутации митохондральных ДНК, контролирующих дыхательную цепь митохондрий, сопровождаются нарушением процессов окислительного фосфорилирования — важнейшего источника энергии для метаболических процессов в клетке.
Клинические проявления
В возрасте до 40 лет пациенты с МЕЛАС поступают с клиникой транзиторной ишемической атаки, а также с эпилепсией, неоднократной рвоты, головной болью, мышечную слабость. У данных пациентов нередко клинически выявляют деменцию.
Молодой возраст и отсутствие факторов риска, характерных для инсульта, помогает задуматься о МЕЛАС.
Лабораторные данные
Лактат ацидоз — увеличения уровня лактата и пирувата.
Данные визуализации
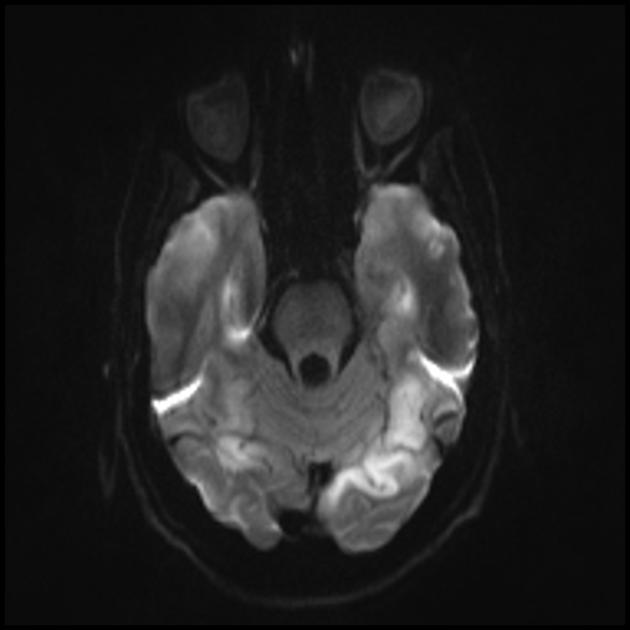
Изменения головного мозга схожи с изменениями при инсульте.
Отличия от инсульта
1) области поражения не совпадают с границами артериальных сосудистых бассейнов.
2) при повторных приступах очаги визуализируются в другой локализации.
+ клинические данные (молодой возраст, отсутствие факторов риска инсульта).
КТ
Множественные гиподенсивные области не соответствующие сосудистому бассейну.
Кальцификация базальных ганглий (наиболее чаще у пожилых пациентов).
Атрофия возникает на фоне регресса и клинического улучшения.
МРТ
Острый инфаркт
Для дифференциации с инсультом используют ADC и DWI (при инсультах ограничение диффузии (цитотоксический отек), а при МЕЛАС диффузия ограничена незначительно, либо без изменений (вазогенный отек).
Вовлечение в патологический процесс субкортикального белого вещества головного мозга.
Ухудшение визуализации четкости контуров извилин и повышение сигнала от них на Т2-взвешенных изображения.
Хронический инфаркт
Изменения могут быть симметричными и ассиметричными.
Фокальная атрофия возникает на фоне регресса и клинического улучшения.
Теменная, затылочная и височная доля головного мозга наиболее чаще поражаются.
МР-спектроскопия
Повышение уровня лактата.



Материалы рассчитаны на врачей-неврологов, терапевтов, общей практики.
Сергей Лихачёв, заведующий, доктор мед. наук, профессор;
Инесса Плешко, ведущий научный сотрудник, кандидат мед. наук, неврологический отдел РНПЦ неврологии и нейрохирургии.
Церебральная аутосомно-доминантная артериопатия с субкортикальными инфарктами и лейкоэнцефалопатией (CADASIL) -прогрессирующее аутосомно-доминантное заболевание, клинические проявления которого включают повторные подкорковые ишемические инсульты, мигрень, субкортикальную деменцию и аффективные нарушения. Распространенность в настоящее время - 1 случай
на 100 000 населения.
В РНПЦ неврологии и нейрохирургии наблюдаются 7 пациентов (в т. ч. 4 женщины) с CADASIL; возраст - от 32 до 68 лет. Их обследовали неврологическим, молекулярно-генетическим методами. Имела место характерная симптоматика; в анамнезе - мигрень, рецидивирующие лакунарные инсульты и аффективные нарушения. МРТ головного мозга позволила обнаружить свойственные CADASIL субкортикальные инфаркты и лейкоэнцефалопатию.
У 2 человек в результате молекулярно-генетической диагностики выявлена гетерозиготная мутация в гене Notch3 на 19-й хромосоме, чем и вызывается CADASIL. Гены Notch кодируют трансмембранные рецепторы, участвующие в онтогенезе клетки. При CADASIL в большинстве случаев определяют миссенс-мутации, из-за которых изменяются структуры трансмембранного белка и нарушаются его функции.
Патогенез CADASIL окончательно не ясен. Считается, что основной фактор - артериопатии с прогрессирующей окклюзией мелких перфорирующих сосудов белого вещества головного мозга (приводит к хронической гипоперфузии). При этом обнаруживают характерные гранулярные осмиофильные включения, вызывающие пролиферацию компонентов базальной мембраны, утолщение средней оболочки и механическое сдавление мелких артерий. В итоге повреждается гематоэнцефалический барьер - развивается отек.
Дополнительный патологический фактор - активация астроцитов вблизи сосудистой стенки. Они высвобождают эндотелии-1, провоцируя вазоконстрикцию и нарушение кровотока.
Состав гранулярных осмиофильных включений неизвестен. Предполагается, что белок Notch3 является одним из их компонентов. При биопсии кожи пациентов с мутацией в гене Notch3 осмиофильные гранулы и дегенерация гладких мышечных клеток могут определяться еще до 20-летнего возраста.
Клиническая диагностика CADASIL:
- отягощенный семейный анамнез;
- развитие первых cимптомов заболевания до 50 лет;
- присутствие двух из сле-дующих симптомов - мигрень, повторные инсульты, нарушения настроения, субкортикальная деменция.
Следует исключить сосудистые факторы риска, этиологически связанные с неврологическими симптомами. МРТ показывает поражение белого вещества полушарий головного мозга и отсутствие кортикальных инфарктов.
Достоверный диагноз «CADASIL» подтверждается положительным результатом молекулярно-генетической диагностики или обнаружением артериопатии с характерными гранулярными осмиофильными включениями при биопсии кожи или мышцы.
Самые частые симптомы CADASIL - преходящие ишемические атаки и ишемические инсульты, наблюдаемые почти у 85% пациентов.
Характеризуются рецидивирующим течением, проявляются классическими синдромами лакунарных инсультов и полной клинической ремиссией через несколько дней или недель.
Вторые по частоте - когнитивные нарушения (отмечаются у 60% пациентов). Могут начинаться в 35 лет, иногда еще до ишемических эпизодов. Приблизительно в 75% случаев при CADASIL развивается деменция. Первый симптом - обычно мигрень; часто возникает до 20 лет и, как правило, предшествует инсультам.
Данные о вовлечении сердца в патологический процесс при CADASIL противоречивые. L. Oberstein et al. (2003 г.) обнаружили, что 25% пациентов с диагностированным CADASIL имели острый инфаркт миокарда в анамнезе или патологию Q-волны на электрокардиограмме. В другом исследовании Cumurciuc et al. (2006 г.) не выявили положительного кардиологического анамнеза у 23 человек с мутацией в гене Notch3.
Клинические проявления CADASIL и микроангиопатии головного мозга иной этиологии схожи - требуется дифференциальная диагностика.
Чтобы своевременно определить CADASIL у пациентов и членов их семей, надо прибегать к молекулярно-генетическим методам и/или гистологическим исследованиям.
Синдром MELAS
Митохондриальная энцефаломиопатия с лактат-ацидозом и инсультоподобными эпизодами (MELAS) - редкое наследственное заболевание, обусловленное патологией митохондриального генома, нарушением энергетического метаболизма и функционирования наиболее энергозависимых органов и тканей (ЦНС, сердечная и скелетные мышцы, глаза, почки, печень, костный мозг, эндокринная система). Широкая вариабельность клинических проявлений MELAS-синдрома и редкая встречаемость предопределяют трудности при диагностике для практического врача.
В РНПЦ неврологии и нейрохирургии наблюдаются 3 пациента (46-летняя женщина и ее сыновья - 24 и 23 лет) с диагностированным синдромом MELAS. Они прошли клинико-неврологическое обследование, молекулярно-генетическую диагностику, МРТ головного мозга.
У всех низкорослость; в анамнезе - симптомы митохондриальной патологии: нейросенсорная тугоухость, мигренеподобные головные боли, плохая переносимость физических нагрузок. Дебют заболевания - генерализованные судорожные приступы. У 2 пациентов первые симптомы проявились до 20 лет; имели место эпилептические припадки, следующие один за другим, эпизоды нарушения зрения с наличием очагов при нейровизуализации в затылочных и височных областях, повышение уровня лактата в крови и цереброспинальной жидкости. У 1 человека выявлено умеренное снижение когнитивных функций; по данным УЗИ сердца - гипертрофическая кардиомиопатия; сахарный диабет.
При молекулярно-генетическом исследовании обнаружены типичные для MELAS мультисистемность поражения, широкая вариабельность и различная степень выраженности клинических проявлений, соответствующие количеству мутантных копий А3243G в тРНКLeu(UUR) гене.
Для MELAS характерны материнский тип наследования, наличие спорадических случаев при возникновении мутации de novo; накопление в клетках - как нормальных, так и мутантных типов - митохондриальной ДНК (гетероплазмия) и случайное распределение при делении между дочерними клетками (митотическая сегрегация). На генетическом уровне причина MELAS-синдрома - гетероплазмическая перестановка 3243A>G в тРНКLeu(UUR) гене (обнаруживается 80% случаев).
Патогенез заболевания пока не изучен. Существуют 2 основные теории - «митохондриальной ангиопатии» и «митохондриальной цитопатии». Известно, что инсультоподобное поражение не соответствует сосудистым зонам и распространяется на окружающие области вследствие сопутствующего вазогенного отека, вызванного длительной эпилептической активностью. Как предполагают, инсультоподобные эпизоды обусловлены нейронной гипервозбудимостью в ограниченном участке мозга. Она возникает из-за митохондриальной дисфункции в эндотелиальных клетках капилляров, или в нейронах, или в астроцитах; деполяризует смежные нейроны, приводя к распространению эпилептической активности.
Кроме того, в промежутках между инсультоподобными эпизодами, по данным однофотонно-эмиссионной компьютерной томографии (SPECT), у пациентов с MELAS наблюдается гипоперфузия коры задней части поясной извилины, что свидетельствует о расстройстве мозговой гемодинамики.
Нарушение окислительного фосфорилирования, разрыв митохондриальной дыхательной цепи способствуют тому, что преобладают катаболический метаболизм и изменения от цикла Кребса к анаэробному гликозу с накоплением лактата. Высокий уровень последнего в ЦНС обычно коррелирует с периодами неврологической симптоматики.
Основные клинические признаки MELAS - инсультоподобные эпизоды, лактат-ацидоз, наличие в мышечных биоптатах «рваных красных волокон». Дополнительными проявлениями могут быть деменция, психозы, эпилептические пароксизмы, мигренеподобные головные боли, атаксия, миопатия, кальцификация базальных ганглиев по данным нейровизуализации, оптическая атрофия, ретинопатия, глухота, диабет, интестинальная псевдообструкция, кардиомиопатия.
Ранний возраст дебюта MELAS - от 5 до 20 лет, однако есть наблюдения и позднего начала - на 5–6-й декадах жизни. Известны случаи, когда синдром запускался после кардиологических нарушений.
Мультисистемность поражения при MELAS осложняет клиническую диагностику.
Наследственный характер заболевания обязывает провести молекулярно-генетические исследования, чтобы поставить точный диагноз
и выявить других пациентов - из числа родственников больного.
Материалы рассчитаны на врачей-неврологов, терапевтов, общей практики.
























1 из 24
Презентация на тему: Синдром MELAS
№ слайда 1

№ слайда 2

Описание слайда:
Синдром MELAS (англ. Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes - «митохондриальная энцефаломиопатия, лактатацидоз, инсультоподобные эпизоды») - прогрессирующее нейродегенеративное заболевание, характеризующееся проявлениями, перечисленными в названии, и сопровождается полиморфной симптоматикой - диабетом, судорогами, снижением слуха, сердечными заболеваниями, низким ростом, эндокринопатиями, непереносимостью физических нагрузок и нейропсихиатрическими отклонениями.
№ слайда 3

Описание слайда:
История Синдром MELAS впервые выделен в нозологически самостоятельную форму S. Pavlakis и соавт. в 1984 г.. Однако есть основания предполагать, что заболевание было описано раньше под названием "семейная полиодистрофия, митохондриальная миопатия, лактацидемия". К 1994 г. в литературе опубликовано 110 наблюдений синдрома MELAS.
№ слайда 4

Описание слайда:
№ слайда 5

Описание слайда:
Этиология, патогенез MELAS относится к митохондриальным заболеваниям. Синдром розвивается в результате точечных мутаций митохондриальной ДНК. Выявлена локализация 3 точечных мутаций, с которыми ассоциирован синдром MELAS: две - в транспортной РНК и одна - в цитохром с- оксидазе.
№ слайда 6

Описание слайда:
№ слайда 7

Описание слайда:
№ слайда 8

Описание слайда:
Патоморфологические изменения Характерным патоморфологическим признаком синдрома MELAS, как и ряда других митохондриальных энцефаломиопатий (синдромов Кернса - Сейр, MERRF и др.), являются "рваные" красные волокна (RRF), которые проявляются в мышечной ткани при модифицированном окраске трихромом по Гомори. Они являются морфологическим субстратом повреждения митохондриальной ДНК и образуются вследствие пролиферации аномальных митохондрий. Красные "рваные" волокна являются следствием мутаций, повреждающих гены транспортной РНК и приводят к нарушению внутримитохондриального синтеза белка. Показано, что такие морфологические характеристики мышечной ткани, как наличие сосудов с высокой активностью сукцинатдегидрогеназы и значительное количество цитохром с-оксидазопозитивних мышечных волокон, является характерной особенностью синдрома MELAS, которые позволяют дифференцировать его от синдромов Кернса-Сейр и MERRF. Одной из наспецифичниших признаков повреждения мозга при данной болезни является наличие старых и новых очагов инфарктов.
№ слайда 9

Описание слайда:
"рваные" красные волокна (RRF)
№ слайда 10

Описание слайда:
Клиническая картина Первые признаки чаще появляются в возрасте 6-10 лет, хотя возможны как более раннее начало заболевания (до 2 лет), так и более поздний (21-40 лет). До появления первых признаков заболевания большинство больных развивается нормально. Начальные клинические проявления: судороги, рецидивирующие головные боли, рвота, анорексия, непереносимость физической нагрузки, психические нарушения, неврологические симптомы (парезы, атаксия и др.).
№ слайда 11

Описание слайда:
Клиническая картина Непереносимость физических нагрузок, после которых ухудшается самочувствие, появляется мышечная слабость, иногда миалгии. Инсультоподобные эпизоды проявляются рецидивирующими приступами головной боли, головокружением, развитием очаговой неврологической симптоматики (парезы, параличи конечностей, черепных нервов), коматозное состояние. Судороги при синдроме MELAS очень вариабельны-фокальные пароксизмы, генерализованные тонико-клонические, миоклонии. Судороги мало чувствительны к противосудорожных терапии.
№ слайда 12

Описание слайда:
Клиническая картина Деменция, обычно, развивается вместе с прогрессированием заболевания, но относительно редко она выступает в роли манифестного симптома. Миопатический симптомокомплекс (мышечная слабость, быстрая утомляемость, иногда гипотрофия). При раннем дебюте заболевания его ход более злокачественный. Так, при дебюте MELAS до 20 лет летальность составляет 30%.
№ слайда 13

Описание слайда:
Основные диагностические критерии: непереносимость физических нагрузок;начало заболевания до 40 лет (чаще до 20 лет);инсультоподибные эпизоды;судороги;"рваные" красные волокна в биоптатах скелетных мышц; лактат-ацидоз;прогрессирующая деменция;миопатический синдром;низкорослость;глухота.
№ слайда 14

Описание слайда:
Дополнительные диагностические критерии: кальцификация базальных ганглиев при компьютерной томографии (КТ) или магнитно - резонансной томографии (МРТ) головного мозга;атаксия;коматозные состояния;атрофия зрительных нервов;пигментный ретинит;синдром Вольфа - Паркинсона – Уайта;сердечная недостаточность;прогрессирующая наружная офтальмоплегия;нарушение проводимости сердца;сахарный диабет.
№ слайда 15

Описание слайда:
Данные лабораторных и функциональных исследований: Характерным признаком заболевания является выявление лактатацидоза в крови и спинномозговой жидкости. У половины больных в ликворе выявляется повышение уровня лактата и белка.Большое значение имеет исследование ферментов дыхательной цепи, чаще выявляются изменения в активности ферментов комплекса I. ЭКГ: могут выявляться нарушения сердечной проводимости, синдром Вольфа-Паркинсона-Уайта. КТ головного мозга: зоны инфарктов чаще в гемисферы, реже в мозжечке, базальных ганглиях. Может наблюдаться кальцинация базальных ганглиев, атрофия коры головного мозга. Церебральная ангиография: увеличение калибра сосудов (артерий, вен, капилляров).
№ слайда 16

Описание слайда:
МРТ головного мозга больной А., Т2взвешенные изображения. а, б – 1е исследование: симметричные очаги повышенной интенсивности сигнала в проекции теменных долей обоих полушарий. в, г – 2е исследование: в проекции височной и теменной долей правого полушария отмечается расширение зоны измененного МР сигнала. В левом полушарии в проекции теменной до ли размеры патологического очага заметно уменьшились.
№ слайда 17

Описание слайда:
КТ головного мозга: в височной доле левого полушария с частичным распространением на теменную долю определяется очаг слабо пониженной плотности (стрелка). В обоих полушариях в области лентикулярных ядер и зрительных бугров определяются очаги повышенной плотности (кальцификаты).
№ слайда 18

Описание слайда:
Исследование церебральной перфузии методом ОФЭКТ у больной А. а–в – исследование 12.11.2003 г. – зона низкой перфузии (синий и зеленый цвета) в височной области правого полушария (а) и высокой перфузии (красный цвет) в затылочных долях (б) и височной доле левого полушария (в). г–е –исследование 18.02.2004 г. – снижение перфузии в левой височной доле (е) по сравнению с предыдущим исследованием.
Описание слайда:
Лечение Симптоматическое.Для коррекции биохимических дефектов используется коэнзим Q10 (80 - 300 мг / сут), витамины К1, К3 - филлохинон (25 мг / сутки) и менадион (до 75 мг / сут), янтарная кислота (до 6 мг / сут), витамин С (2-4 г / сут) и другие витамины (рибофлавин, тиамин, никотинамид). Известно, что коэнзим Q10 в физиологических условиях переносит электроны от комплексов I и II к комплексу III и содействует тем самым стабилизации дыхательной цепи, уменьшению уровня лактата и пирувата. Витамины К1 и К3, очевидно, способны выполнять функцию транспорта электронов на уровне I и III комплексов. Янтарная кислота обеспечивает передачу электронов II комплекса. Витамин С рассматривается как донор электронов IV комплекса, а также как важный антиоксидант. Кроме аскорбиновой кислоты, для предупреждения кислородно - радикального повреждения митохондриальных мембран назначается витамин Е (300 - 500 мг / сутки).
№ слайда 21

Описание слайда:
Лечение С целью стимуляции синтеза АТФ предлагается использовать идебенон (90 - 180 мг / сут), который обладает свойством усиливать энергетический метаболизм в мозговой ткани. Введение витаминов рибофлавина (100 мг / сутки) и никотинамида (до 1 г / сут) - предшественников коэнзимов НАД и ФАД, принимающих активное участие в окислительных процессах, также способствуют улучшению энергетической продукции митохондрий. В связи со вторичным карнитиновим дефицитом, больным назначают L-карнитин (до 100 мг / сутки). С целью снижения уровня лактата в крови и спинно-мозговой жидкости используется дихлорацетата натрия (25-100 мкг / кг).С помощью лабораторных тестов нужно проверить возможные нарушения функций эндокринной системы (сахарный диабет, гипопаратиреоз) и сердечно-сосудистой системы (блокада). При выявлении нарушений проводится их медикаментозная коррекция.
№ слайда 24

Описание слайда:
Вестник АГИУВ, спецвыпуск, 2013г. удк 616.8-007: 616.853.3
дебют сидромл melas с фебрильными судорогами
(случай из практики)
т. о. мусабекова, А. и. хамзина
Кыргызско-Российский Славянский Университет, Кафедра неврологии и нейрохирургии, г. Бишкек, Кыргызстан
Фебрильные судороги (ФС) известны со времен античности. Еще Гиппократ писал, что ФС наиболее часто возникают у детей первых 7 лет жизни и гораздо реже - у более старших детей и у взрослых . Но впервые термин «фебрильные судороги» применил в 1904 году B. Hochsinge для обозначения судорожных пароксизмов, развивающихся в детском возрасте на фоне лихорадки. В настоящее время предпочтительнее говорить о фебрильных приступах (ФП), а не ФС, так как в клинической картине данного состояния могут наблюдаться не только судорожные, но и бессудорожные пароксизмы 2]. По определению ILAE от 1993 года, ФП - это приступы, отмечающиеся у детей в возрасте старше 1 месяца, связанные с фебрильным заболеванием, не вызванным инфекцией ЦНС; без предшествующих судорог в неонатальном периоде и неспровоцированных приступов, а также не соответствующие критериям других острых симптоматических приступов. Согласно проекту классификации 2001 года, ФП отнесены в группу состояний, которые не требуют обязательного диагноза эпилепсии . Таким образом ФП определяются как эпизод эпилептических приступов, возникающих у детей в возрасте от 6 мес. до 5 лет при повышении температуры в период вирусного или бактериального заболевания, не связанного с нейроинфекцией и метаболическими нарушениями. Истинные ФП следует отличать от фебрильно провоцируемых приступов, которые могут входить в структуру ряда форм эпилепсии, например при синдроме Драве. В редких случаях ФП могут быть первым симптомом митохондриальных заболеваний у детей .
синдром MELAS (митохондриальная энцефало-ми-опатия с лактат-ацидозом и инсультоподобными эпизодами) был впервые выделен в отдельную нозологическую форму S. Pavlakis et al. только в 1984 г. . Заболевание относится к группе митохондриальных болезней, связанных с точковыми мутациями митохондриальной ДНК, в результате которых происходит нарушение энергопродукции в митохондриальной дыхательной цепи . Известно что точечные мутации могут возникать во многих генах (MTTL1, MTTQ, MTTH, MTTK, MTTS1, MTND1, MTND5, MTND6, MTTS2), наследоваться по материнской линии . Распространённость синдрома MELAS сложно оценить из-за разнообразия проявлений и связанной с этим трудностью диагностики. к 2000 году было опубликовано более 120 наблюдений заболевания . Кардинальными симптомами при MELAS-синдроме являются: непереносимость физических нагрузок, инсультоподобные эпизоды, судороги, «рваные красные» волокна в биоптатах мышечной ткани, лактат-ацидоз и дебют заболевания в возрасте до 40 лет . Синдром MELAS следует дифференцировать с другими митохондриальными заболеваниями: синдромом Кернса-Сейера и MERRF.
Ниже приводится наше собственное наблюдение больной П. 2003-го года рождения, проживающей в городе Бишкек. Ребенок обратился к нам в центр клиники МЕБИ ЛТД города Бишкек в весной 2013 года с жалобами на тонико-клонические судороги в ногах и руках длительностью до 2 минут, протекающие с потерей сознания и развивающиеся
только на фоне повышения температуры тела выше 37 С°, а также на появление сложности в усвоении школьного материала, снижение памяти, повышенную утомляемость и мышечную слабость, неловкость при ходьбе.
Дебют заболевания у девочки отмечался в возрасте 6-ти месяцев с генерализованного тонико-клонического приступа продолжительностью до 1 минуты на фоне повышения температуры тела до 38 С°, после которого была госпитализирована в Республиканскую инфекционную больницу города Бишкек, где исключили нейроинфекцию. В последующем ФП возникали каждый раз при повышении температуры тела выше 37 С°. В возрасте 1 год при обращении в Национальный центр педиатрии и детской хирургии КР, было проведено МРТ головного мозга, ЭЭГ, где патологии не выявлено, был назначен депакина в дозе 20 мг/кг/сут. Тем не менее ФП продолжались на фоне приема противосудорожного препарата. В возрасте 5-ти лет самостоятельно обратились в Республиканскую детскуюклиническую больницу (РДКБ) города Москвы, где было повторно проведено МРТ головного мозга и видео - ЭЭГ -мониторинг (ВЭМ) дневного сна, где вновь патологии не выявлено. Врачами РДКБ был выставлен диагноз криптогенная эпилепсия, рекомендовано повысить дозу депакина до 25 мг/кг/сут. Первые три класса в общеобразовательной школе закончила на оценки «4» и «5». С 9-ти лет мама стала отмечать постепенное нарастание у ребенка быстрой утомляемости после физических нагрузок, появление сложности в усвоении школьного материала, встал вопрос о переводе ребенка в специализированное школьное учреждение для детей с умственной отсталостью. ФП продолжали беспокоить ребенка и после 7-ми лет на фоне приема депакин хроно в дозе 25 мг/кг/сут.
Из анамнеза жизни: ребенок от первой беременности протекавшей на фоне легкого токсикоза в первом триместре, юыл эпизод ОРВИ без температуры в сроке 5 мес. Роды в срок, самостоятельные в головном предлежании, по шкале Апгар 7/8 баллов, ВПР - 3340 гр., рост - 52 см. Раннее развитие ребенка соответствовало возрастной норме.
На момент осмотра в в клинике числа возрасте 10-ти лет со стороны черепно-мозговых нервов отмечается незначительная девиация языка вправо, миопатический синдром в руках и ногах в виде гипотонии, легкой гипотрофии проксимальных отделов рук и ног со снижением мышечной силы до 4 баллов, снижении сухожильных рефлексов, а также легкое пошатывание в позе Ромберга и неловкость при выполнении пальце-носовой и коленно-пяточной проб, снижение кратковременной памяти и внимания.
Дополнительные обследования: Нв 112 г/л, эритроциты 3,5 *10"12/л, печеночные тесты, общий белок, сахар, креатинин в пределах нормы.
Продолжение ФП после 5-ти лет с развитием резистентности к вальпроатам, присоединение миопатического синдрома и снижение когнитивных функций позволило высказать предположение о возможности наличия у больной митохондриальной патологии, а именно синдрома MELAS, что потребова-
ло проведения ряда дополнительных исследований. При электронейромиографии проведенной в НЦП и ДХ выявлен первично-мышечный тип поражения в виде уменьшения длительности потенциала двигательных единиц на 30-35% и снижения их амплитуды с нормальной скоростью проведения по периферическим нервам. При повтороном ВЭМ патологии не выявлено. В SVS лаборатории имени В.М. Савинова города Алматы была определена концентрация депакина в крови до приема препарата- 86, 98 нг/мл и через 2 часа после приема препарата -113, 61 нг/мл при норме 50-100 нг/мл. В Приват клинике города Алматы было определено содержание молочной кислоты натощак - 3,1 ммоль/л, при норме до 1,7ммоль/л. Был выставлен предварительный диагноз синдром MELAS, депакин заменен на кеппру, введен коэнзим Q10, карнитин, витамины группы В, витамин Е, диета с ограничением приема углеводов, рекомендовано проведение генетического обследования.
Таким образом, в представленном нами клиническом случае у ребенка наблюдались простые ФП, для профилактического лечения которых был назначен депакин с длительным приемом на протяжении нескольких лет несмотря на наличие фармакорезистентности. Принимая во внимание отсутствие четких доказательств эффективности профилактического применения антиконвульсантов у детей с ФП, назначение депакина в данном случае было нецелесообразно. Так по литературным данным длительное применение депакина и барбитуратов существенно усугубляет течение митохондриальных заболеваний, порой приводя к прогрессированию патологического процесса , что и произошло в нашем клиническом случае.
список литературы:
1. Гузева В.И. Специальные синдромы (ситуационно-обусловленные приступы) / В.И. Гузева // Эпилепсия и неэпилептические пароксизмальные состояния у детей.-М.: МИА, 2007.- С. 443- 457
2. Мухин К.Ю. Фебрильные судороги /А.С. Петрухин
// Неврология детского возраста. - М.: Медицина, 2004.-С.664-668.
3. Никанорова М.Ю., Темин П.А., Кобринский Б.А. Фебрильные судороги / П.А. Темина, М.Ю. Никаноровой // Эпилепсия и судорожные синдромы у детей.- М.: Медицина, 1999.- С. 169- 195.
4. Основные методы лечения детей, страдающих митохондриальными заболеваниями: Методич. указания.-М., 2001.
5. Темин П.А. и др. //Неврол. журн.- 1998. № 2.- С. 43.
6. Яхно Н.Н. и др. //Неврол. журн. 1998.- № 5.- С. 14.
7. Ban S. et al. //Acta Pathol. Jpn.- 1992. -Vol. 42. -P. 818.
8. Hirano M., Pavlakis S.G. // J. Clin. Neurol. -1994.-Vol. 9. -P. 4.
9. ILAE Commission report: glossary of descriptive terminology for ictal semiology: report of the ILAE Task Force on Classification and Terminology /Epilepsia.- 2001.- Vol. 42. -P.1212-1218.
10. ILAE. Guidelines for epidemiologic studies on epilepsy /Epilepsia.- 1993.- Vol.34.- P. 592-596.
11. Pavlakis S.G. et al. //Ann. Neurol.-1984. -Vol. 16.-P. 481.
12. Sciacco M. et al. // J. Neurol. -2001. -V 248. -P. 778.
Фебрильные судороги нередко могут быть первым симптомом митохондриальных заболеваний у детей, что значительно затрудняет своевременную диагностику и начало патогенетического лечения болезни, а порой обуславливает применение препаратов ухудшающих течение и прогноз заболевания.
ключевые слова: судороги, терапия.
Febrile seizures can often be the first symptom of mitochondrial disease in children, which greatly complicates the timely diagnosis and early treatment, and sometimes causes the use of drugs worsen the course and prognosis of the disease.
Keywords: convulsions, therapy.
удк 616.831-005.4
СЛУЧАИ ИШЕМИЧЕСКОГО ИНСУЛЬТА КАК ПРОЯВЛЕНИЯ МИТОХОНДРИАЛЬНОЙ ЭНЦЕФАЛОПАТИИ У БОЛЬНОГО МОЛОДОГО
ВОЗРАСТА
Карбозова К.З., Луценко И. Л.
Кафедра неврологии с курсом медицинской генетики, Кыргызская Государственная Медицинская Академия имени И.К. Ахунбаева,
г. Бишкек, Кыргызстан
Распространенность инсульта в молодом возрасте (до 45 лет) составляет от 2,5 до 10% всех случаев нарушений мозгового кровообращения и продолжает увеличиваться . У пациентов молодого возраста наиболее частыми причинами развития ишемических сосудистых нарушений являются: аномалии цереброваскулярной системы, диссекция, кардиальная патология мигрень, дефекты коагуляции, АФЛС, .
За 5 последних месяцев в отделении неврологии №1 Национального госпиталя при Министерстве Здравоохранения Кыргызской Республики (НГМЗКР) получало лечение 608 больных. Произведен анализ 46 (7.5) историй болезни пациентов, перенесших ишемический инсульт, из них 4 (8.7%) молодого возраста (до 45 лет по ВОЗ). В таблице 1 приведены подтипы ишемического инсульта.
Представляем историю болезни пациента, госпитализированного с первоначальным диагнозом острое нарушение мозгового кровообращения (оНмК), у которого истинную природу заболевания удалось установить только при динамическом наблюдении и специальном дополнительном обследовании.
Таблица 1.
Подтип инсульта Число больных в %
Атеротромботиче ский 32 69,5
Лакунарный 6 13,04
Гемореологический 3 6,5
Кардио эмболиче ский 3 6,5
Митохондриальный 1 2,2