Encefalomiopatie mitocondrială cu acidoză lactică și episoade de melas asemănătoare accidentului vascular cerebral. Boli rare Debutul sindromului melas la vârsta adultă Observare
Cuvinte cheie
SINDROM MELAS / EPILEPSIE / EPILEPSIE / CLINICAadnotare articol științific despre medicina clinică, autor al lucrării științifice - Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova S.V., Chadaev V.A.
Sindromul MELAS este o boală determinată genetic din grupa bolilor mitocondriale, definită ca encefalomiopatie mitocondrială cu acidoză lactică și episoade asemănătoare AVC (encefalomiopatie mitocondrială, acidoză lactică cu episoade asemănătoare AVC). Toate organele și țesuturile sunt implicate în procesul patologic, dar sistemul muscular și nervos suferă într-o măsură mai mare. Boala se dezvoltă cel mai adesea între 6 și 10 ani. Cursul bolii este progresiv. În cele mai multe cazuri, boala se manifestă prin convulsii epileptice, dureri de cap recurente, vărsături și anorexie. Epilepsia este o manifestare clinică importantă a sindromului MELAS. Crizele epileptice sunt primul simptom recunoscut în encefalopatiile mitocondriale (EM) în 53% din cazuri. În MELAS, epilepsia occipitală este cea mai frecventă. Odată cu progresia bolii, se observă rezistența epilepsiei la terapie, adesea cu un curs de stare. Sunt descrise cazuri de transformare în epilepsia lui Kozhevnikov. Prezentăm istoricul unui pacient cu diagnostic de sindrom MELAS verificat în timpul vieții.
Subiecte asemănătoare lucrări științifice în medicina clinică, autor al lucrărilor științifice - Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova S.V., Chadaev V.A.
-
Encefalopatie mitocondrială cu episoade asemănătoare accidentului vascular cerebral și acidoză lactică (sindrom melas): criterii de diagnostic, caracteristici ale crizelor epileptice și abordări ale tratamentului pe exemplul unui caz clinic
2017 / Yamin M.A., Chernikova I.V., Araslanova L.V., Shevkun P.A. -
Accident vascular cerebral în bolile mitocondriale
2012 / Pizova N.V. -
Epilepsia la copiii cu boli mitocondriale: caracteristici de diagnostic și tratament
2012 / Zavadenko N. N., Kholin A. A. -
Tulburări neurologice în encefalomiopatia mitocondrială - acidoză lactică cu episoade asemănătoare accidentului vascular cerebral (sindrom MELAS)
2012 / Kharlamov Dmitry Alekseevich, Krapivkin Alexey Igorevich, Sukhorukov Vladimir Sergeevich, Kuftina Lyudmila Andreevna, Groznova Olga Sergeevna -
Sindromul Melas ca o cauză neobișnuită a hipoparatiroidismului: un caz clinic
2018 / Umyarova Dilyara Shamilevna, Grebennikova Tatyana Alekseevna, Zenkova Tatyana Stanislavovna, Sorkina Ekaterina Leonidovna, Zhanna Belaya -
Episoade asemănătoare accidentului vascular cerebral în encefalomiopatia mitocondrială cu acidoză lactică
2010 / Kalashnikova Lyudmila Andreevna, Dobrynina L. A., Sakharova A. V., Chaikovskaya R. P., Mir-kasimov M. F., Konovalov R. N., Shabalina A. A., Kostyreva M. V., Gnezditsky V.V., Protsky -
Citopatii mitocondriale: melas și sindroame MIDD. Un defect genetic, fenotipuri clinice diferite
2017 / Muranova A.V., Strokov I.A. -
Epilepsie occipitală benignă a copilăriei cu debut precoce (sindrom Panayotopoulos). Descrierea cazului clinic
2015 / Matyuk Yu.V., Kotov A.S., Borisova M.N., Panteleeva M.V., Shatalin A.V. -
Polimorfismul manifestărilor clinice ale encefalomiopatiei mitocondriale progresive asociate cu mutația genei POLG1
2016 / Yablonskaya M.I., Nikolaeva E.A., Shatalov P.A., Kharabadze M.N. -
Valoarea diagnostică a studiului activității citochimice a enzimelor în bolile mitocondriale ereditare
2017 / Kazantseva I.A., Kotov S.V., Borodataya E.V., Sidorova O.P., Kotov A.S.
EPILEPSIA ÎN SINDROMUL MELAS
Sindromul MELAS este o boală determinată genetic a grupului mitocondrial, definită ca encefalomiopatie mitocondrială, acidoză lactică cu episoade asemănătoare accidentului vascular cerebral. Procesul patologic implică toate organele și țesuturile, dar este mai ales advers pentru sistemele musculare și nervoase. Boala este cel mai frecventă la copiii cu vârsta cuprinsă între 6 și 10 ani. Evoluția clinică este progresivă. În majoritatea cazurilor boala se manifestă prin convulsii epileptice, dureri de cap recidivante, vărsături, anorexie. Prezentarea clinică importantă a sindromului MELAS este epilepsia. Crizele de epilepsie reprezintă simptomul de diagnostic inițial al encefalopatiilor mitocondriale (EM) în 53% din cazuri. Epilepsia occipitală este cea mai frecventă în sindromul MELAS. Pe măsură ce boala progresează, se observă rezistența epilepsiei la tratament, adesea cu apariția statusului epilepticus. Sunt descrise unele cazuri de transformare în epilepsia Kozhevnikov.Se prezintă istoricul unui pacient cu un diagnostic verificat în viață de sindrom MELAS.
Textul lucrării științifice pe tema „Epilepsia în sindromul melas”
VOLUM IV NUMĂRUL 3 2009
EPILEPSIA CU SINDROMUL MELAS
K.Yu. Mukhin1, M.B. Mironov1, N.V. Nikiforova1, C.B. Mihailova2, VA. Chadaev1, AA. Alikhanov1-2, B.N. Ryzhkov1, A.S. Petruhin1
EPILEPSIA ÎN SINDROMUL MELAS
KYu. Mukhin1, M.B. Mironov1, N.V. Nikiforova1, S.V. Mihailova2, UA. Chadaev1, AA. Alikhanov1-2, B.N. Ryzkov1 AS. Petruhin1
1 - Departamentul de Neurologie și Neurochirurgie, Facultatea de Pediatrie, Instituția de Învățământ de Stat de Învățământ Profesional Superior, Universitatea de Medicină de Stat Rusă din Roszdrav
2 - Spitalul Clinic de Copii din Rusia
Sindromul MELAS este o boală determinată genetic din grupa bolilor mitocondriale, definită ca encefalomiopatie mitocondrială cu acidoză lactică și episoade asemănătoare AVC (encefalomiopatie mitocondrială, acid lactic cu episoade asemănătoare accidentului vascular cerebral). Toate organele și țesuturile sunt implicate în procesul patologic, dar sistemul muscular și nervos suferă într-o măsură mai mare. Boala se dezvoltă cel mai adesea între 6 și 10 ani. Cursul bolii este progresiv. În cele mai multe cazuri, boala se manifestă prin convulsii epileptice, dureri de cap recurente, vărsături și anorexie. Epilepsia este o manifestare clinică importantă a sindromului MELAs. Crizele epileptice sunt primul simptom recunoscut în encefalopatiile mitocondriale (EM) în 53% din cazuri. În MELAS, epilepsia occipitală este cea mai frecventă. Odată cu progresia bolii, se observă rezistența epilepsiei la terapie, adesea cu un curs de stare. Sunt descrise cazuri de transformare în epilepsia lui Kozhevnikov. Prezentăm istoricul unui pacient cu diagnostic de sindrom MELAS verificat în timpul vieții.
Cuvinte cheie: sindrom MELAS, epilepsie, clinică, diagnostic, tratament.
Sindromul MELAS este o boală determinată genetic a grupului mitocondrial, definită ca encefalomiopatie mitocondrială, acidoză lactică cu episoade asemănătoare accidentului vascular cerebral. Procesul patologic implică toate organele și țesuturile, dar este mai ales advers pentru sistemele musculare și nervoase. Boala este cel mai frecventă la copiii cu vârsta cuprinsă între 6 și 10 ani. Evoluția clinică este progresivă. În majoritatea cazurilor boala se manifestă prin convulsii epileptice, dureri de cap recidivante, vărsături, anorexie. Prezentarea clinică importantă a sindromului MELAS este epilepsia. Crizele de epilepsie reprezintă simptomul de diagnostic inițial al encefalopatiilor mitocondriale (EM) în 53% din cazuri. Epilepsia occipitală este cea mai frecventă în sindromul MELAS. Pe măsură ce boala progresează, se observă rezistența epilepsiei la tratament, adesea cu apariția statusului epilepticus. Sunt descrise unele cazuri de transformare în epilepsie Kozhevnikov.Se prezintă istoricul unui pacient cu un diagnostic verificat în viață de sindrom MELAS.
Cuvinte cheie: sindrom MELAS, epilepsie, tablou clinic, diagnostic, tratament.
Sindromul MELAS este o boală determinată genetic din grupa bolilor mitocondriale, definită ca encefalomiopatie mitocondrială cu acidoză lactică și episoade asemănătoare AVC (encefalomiopatie mitocondrială, acidoză lactică cu episoade asemănătoare AVC).
Sindromul MELAS a fost identificat pentru prima dată ca formă nosologică independentă de S. Pavlakis et al. în 1984 . Cu toate acestea, o serie de autori sugerează că boala a fost descrisă mai devreme sub denumirea de „poliodistrofie familială, miopatie mitocondrială, acidemia lactică”.
Prevalența în populație nu a fost stabilită. Până în 2000, au fost publicate peste 120 de observații ale sindromului MELAS, inclusiv în presa internă.
Sindromul MELAS în 25% din cazuri este moștenit matern cu risc ridicat, dar la 56-75% dintre pacienți istoricul familial nu este împovărat. Boala este asociată cu mutații în genele ADN mitocondrial care codifică subunități ale complexelor lanțului respirator și genele ARN de transport (MT-ND1, MT-ND5, MT-TH, MT-TL1 și MT-TV). În 80-90% din cazurile de sindrom MELAS, boala se bazează pe o mutație punctuală a genei MT-TL1 care codifică ARN de transfer de leucină. Cu această mutație, nucleotida adenină este înlocuită cu guanină la poziția 3243 (A3243G), care perturbă sinteza tuturor proteinelor din mitocondrii.
Toate organele și țesuturile sunt implicate în procesul patologic, dar sistemul muscular și nervos suferă într-o măsură mai mare.
Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova C.V., Chadaev V.A., Alikhanov A.A., Ryzhkov BN., Petrukhin A.S.
Epilepsia în Sindromul MELAS Rus. zhur. det. Neur.: vol. IV, nr. 3, 2009.
ARTICOLE ORIGINALE
subiectele ca fiind cele mai volatile. Severitatea manifestărilor clinice depinde de efectul de prag (vârstă, nevoile energetice ale țesuturilor), de controlul genelor nucleare asupra sintezei lanțului respirator, heteroplasmie (conținut diferit de molecule de ADNmt mutante în țesuturi). S-a demonstrat că la pacienții cu sindrom MELAS, conținutul de ADNmt mutant în diferite țesuturi este de 93-96%. La membrii familiei probanda, ADNmt mutant este de asemenea detectat în țesuturi, dar conținutul său este semnificativ mai mic: 62-89% în forma ștearsă a bolii, de la 28 la 89% în absența semnelor clinice ale sindromului.
Boala se dezvoltă cel mai adesea la vârsta de 6 până la 10 ani, dar există cazuri de debut mai devreme (până la doi ani) sau mai târziu - de la 21 la 40 de ani. Înainte de debutul bolii, 90-100% dintre pacienți se dezvoltă normal. Cursul bolii este progresiv, mai malign cu debut precoce.
În cele mai multe cazuri, boala se manifestă prin convulsii epileptice, dureri de cap recurente, vărsături și anorexie. De asemenea, ar trebui să acordați atenție intoleranței la activitatea fizică sub formă de deteriorare a sănătății și apariția slăbiciunii musculare. Complexul de simptome miopatice se manifestă prin intoleranță la efort, slăbiciune musculară, oboseală și uneori hipotrofie musculară.
Pe măsură ce boala progresează, demența se dezvoltă de obicei. Simptomele precum ataxia cerebeloasă, surditatea neurosenzorială și polineuropatia periferică sunt mai puțin frecvente.
Episoadele asemănătoare accidentului vascular cerebral sunt caracteristice, care se pot manifesta prin atacuri recurente de cefalee, amețeli, dezvoltarea simptomelor neurologice focale (pareză, hemianopsie) și comă. Aceste episoade acute sunt adesea declanșate de febră sau infecții intercurente. Aceste manifestări pot avea o regresie destul de rapidă (de la câteva ore la câteva săptămâni), precum și o tendință de recidivă.
Epilepsia este o manifestare clinică importantă care apare adesea în stadiile incipiente ale MELAS. Acest
adesea cea mai evidentă manifestare neurologică, mai ales în encefalopatia mitocondrială atipică (EM). Crizele epileptice sunt primul simptom recunoscut în encefalopatiile mitocondriale (EM) în 53% din cazuri.
În MELAS, epilepsia occipitală (SE) este cea mai frecventă. Caracterizat prin convulsii focale cu originea în lobii occipitali. Convulsiile sunt adesea asociate cu simptome neurologice tranzitorii sau persistente, cum ar fi pierderea câmpului vizual.
Crizele care provin din cortexul occipital sunt împărțite în funcție de manifestările lor în senzații subiective (aura) și simptome detectabile clinic, de regulă, cu o componentă motorie. Aurele epileptice care emană din lobul occipital includ halucinații vizuale simple și complexe, amauroza. Cele mai tipice crize caracteristice SE sunt halucinațiile vizuale simple, care se pot manifesta ca simptome pozitive (blițuri, pete de lumină) și negative (scotom, hemianopsie). Cel mai adesea, halucinațiile vizuale sunt descrise ca o pată sau pete de lumină, fie constante, fie intermitente. De regulă, pata este albă cu o nuanță verzuie. De asemenea, halucinațiile pot fi multicolore sau monocromatice. Halucinațiile apar de obicei în câmpurile vizuale contralaterale la focarul de excitație în cortexul occipital cu răspândire ulterioară. Cu toate acestea, trebuie remarcat faptul că în plângerile pacienților, aura vizuală nu este adesea detectată.
Halucinațiile vizuale complexe sunt observate atunci când excitația epileptică se extinde în regiunile occipito-temporale sau occipito-parietale. Halucinațiile vizuale complexe pot apărea sub formă de oameni, obiecte animale sau scene, pot fi familiare sau necunoscute, plăcute sau terifiante, înspăimântătoare, simple sau grotești, pot fi statice sau se pot mișca în plan orizontal și pot dispărea. De regulă, ele sunt un simptom terminal înainte de dezvoltarea unui atac motor; poate fi primul simptom ictal, dar mai des apar ulterior
VOLUM IV NUMĂRUL 3 2009
halucinații de bază.
Ictal ama vrosis este un tip special de convulsii, extrem de greu de diagnosticat, care emană din cortexul occipital. Potrivit multor autori, acesta este același simptom frecvent al iritației lobului occipital, precum și al halucinațiilor vizuale, dar adesea rămâne nerecunoscut. De obicei, pacienții nu disting acest simptom separat în structura atacului. Pierderea vederii apare bilateral cu pierderea câmpurilor laterale. Posibilă hemianopie omonimă contralaterală la focarul atacului. Senzațiile pacienților sunt descrise de ei ca întunecare în ochi, „întuneric alb”, percepție afectată a culorilor. Poate un curs de statut cu formarea așa-numitului status epilepticus amauroticus.
Crizele occipitale se pot prezenta cu simptome autonome. Acestea includ migrenă, amețeli, greață și vărsături. Un simptom comun este durerea de cap asemănătoare migrenei după atac.
Manifestările clinice ale convulsiilor care apar limitat în cortexul occipital sunt caracterizate prin deviația ochilor în lateral. Deviația ochilor poate fi observată împreună cu abaterea capului în lateral. În cele mai multe cazuri, se observă deviația ochilor către focalizarea contralaterală. Cu toate acestea, sunt descrise cazuri când se observă abducția ochilor spre focalizare. De asemenea, una dintre caracteristicile crizelor „occipitale” este distribuția instantanee a descărcării în părțile anterioare ale creierului, în timp ce tabloul clinic, de regulă, este dominat de o componentă motorie pronunțată. Sunt posibile crize tonice, tonico-clonice (atât hemiconvulsive, cât și secundar generalizate), automotorii. În acest sens, este important să se identifice simptomele clinice inițiale - o oprire nemotivată și bruscă a privirii, privire la obiecte inexistente, un zâmbet nerezonabil, manifestări vegetative și, în mod necesar, documentarea zonei ictogene primare folosind metoda VEM.
Odată cu progresia bolii, se observă rezistența epilepsiei la terapie, adesea cu un curs de stare. Sunt descrise cazuri de transformare în epilepsie Kozhevnikov. O serie de auto-
Rov descrie posibilitatea statusului epilepticus ca primul simptom la pacienții cu MELAS fără antecedente de crize epileptice anterioare. Ribacoba R. şi colab. descriu în publicația lor 4 cazuri de dezvoltare a epilepsiei parțiale continue cu convulsii motorii focale, care a fost precedată în anamneză de episoade de cefalee migrenoasă. Miyazaki M. şi colab. a arătat posibilitatea continuării mioclonului focal în epilepsia parțială continuă la pacienții cu MELAS. Araki T. şi colab. a observat un pacient la vârsta de 37 de ani cu stare epileptică de convulsii focale sub formă de fluctuații ale conștienței, hemianopsie omonimă în combinație cu episoade paroxistice de abatere a ochiului în lateral. EEG a înregistrat modele EEG continue de convulsii localizate în regiunea occipitală. La pacienții adulți cu MELAS, există o predominanță a crizelor focale motorii, dar EEG-ul arată o predominanță a activității epileptiforme multiregionale în regiunile occipitale.
Activitatea epileptiformă se înregistrează în 71% din cazuri după debutul crizelor. Un studiu electroencefalografic al pacienților cu sindrom MELAS se caracterizează prin activitate epileptiformă în regiunile occipitale. O serie de autori asociază apariția tulburărilor epileptiforme regionale cu accidente vasculare cerebrale. Potrivit studiului lui Fujimoto S., în perioada acută (adică, în decurs de 5 zile după un episod asemănător unui accident vascular cerebral), majoritatea pacienților examinați cu sindrom MELAS au avut unde delta regionale de amplitudine mare în combinație cu polispikes. Autorii propun să considere acest model ca fiind patognomonic pentru episoadele asemănătoare accidentului vascular cerebral. Pe lângă regiunile occipitale, activitatea epileptiformă se poate răspândi în regiunile temporale, bifrontal și, de asemenea, bilateral în regiunile posterioare cu distribuție difuză. Poate apariția unui răspuns fotoparoxistic în timpul fotostimularii ritmice.
Semnul principal de laborator este creșterea nivelului de lactat din sânge.
ARTICOLE ORIGINALE
cu peste 2,0 mmol/l, ceea ce duce la dezvoltarea acidozei lactice.
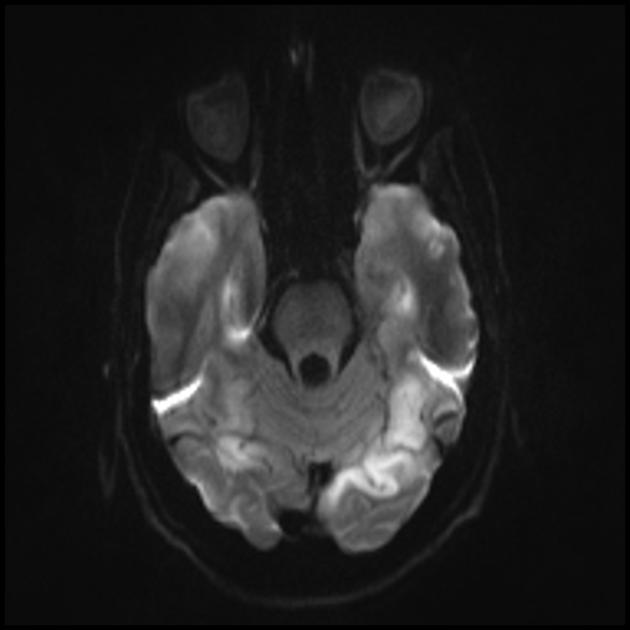
Un RMN al creierului în stadiile incipiente ale bolii poate fi neremarcabil, chiar dacă apare epilepsia. Metodele de neuroimagistică relevă zone de infarct în emisferele cerebrale (80%), mai rar în cerebel și ganglionii bazali. Poate exista și calcificarea ganglionilor bazali, atrofia cortexului cerebral. Într-un studiu de emisie de fotoni, acumularea izotopului este detectată cu 3-16 zile înainte de apariția zonei de infarct (scăderea semnalului izotopului) pe o tomogramă computerizată a creierului. RMN-ul creierului arată leziuni localizate predominant în lobii occipitali, care pot fi tranzitorii. Cortexul occipital este afectat predominant, substanța albă este afectată într-o măsură mai mică. Pe imaginile ponderate T2, leziunile cerebrale din MELA apar ca zone cu intensitate crescută a semnalului. Zonele hiperintense tranzitorii sunt asociate de o serie de autori cu edem vascular reversibil.
Angiografia de obicei nu evidențiază anomalii vasculare. RMN-ul ponderat prin difuzie demonstrează modificări asociate cu edem vasogenic.
Histopatologie: Biopsia musculară evidențiază fibre cu „margini roșii” rupte. Autopsia creierului se caracterizează printr-o combinație de focare vechi și noi de infarcte, precum și atrofie a cortexului cu focare focale de necroză.
În prezent, terapia este de susținere. Direcția principală de tratament este îmbunătățirea echilibrului energetic al mitocondriilor și al lanțului respirator. Se aplica coenzima p10 (80-300 mg/zi), vitaminele K1 si KZ (25 mg/zi), acid succinic (pana la 6 g/zi), vitamina C (2-4 g/zi), riboflavina (100 mg/zi). zi) și nicotinamidă (până la 1 g/zi). În legătură cu dezvoltarea deficitului secundar de carnitină, pacienților li se prescrie L-carnitină (până la 100 mg/kg/zi). Vitamina E (300-500 mg/zi) și vitamina C (2-4 mg/zi) sunt folosite ca terapie antioxidantă.
Nu există regimuri de terapie antiepileptică general acceptate pentru MELA. O serie de autori propun să excludă medicamentele care pot inhiba metabolismul energetic (barbiturice, medicamente cu acid valproic; precum și unele medicamente din alte grupe, de exemplu, cloramfenicol). Literatura descrie mai multe cazuri izolate de agravare a convulsiilor cu utilizarea acidului valproic în sindromul MELA cu mutația A3243C. Principalele FAE în tratamentul epilepsiei în sindromul MELA sunt considerate a fi tegretol (sau trileptal), topamax, keppra în doze terapeutice medii. Terapia selectată în mod corespunzător duce la o scădere semnificativă a frecvenței crizelor convulsive generalizate secundare. Cu toate acestea, crizele cu funcții vegetativ-viscerale și vizuale afectate sunt de obicei rezistente la tratament. În stadiul terminal al bolii, frecvența crizelor epileptice poate scădea.
Iată istoricul unui pacient cu un diagnostic de sindrom MELAY verificat în timpul vieții.
Pacientul Ch.A., în vârstă de 11 ani, a fost observat la Centrul de Neurologie și Epilepsie Pediatrică. La internare, s-au făcut plângeri de pierderea treptată a abilităților de vorbire, o tulburare pronunțată a mersului cu refuzul de a merge, o scădere semnificativă a vederii, capricios și comportament negativ; atacuri în serie zilnice sub formă de zvâcniri ale mușchilor feței, mușchilor extremităților superioare și inferioare, precum și episoade pe termen scurt de pierdere a vederii.
Debutul bolii a fost observat la vârsta de 5 ani și 9 luni. Pentru prima dată, pe fondul sănătății complete, la adormire, a apărut o durere de cap severă, halucinații vizuale simple („raza galbenă”), urmate de o întoarcere violentă a ochilor și a capului în lateral și dezvoltarea unui generalizat. convulsii tonico-clonice, după care s-au observat vărsături. Dupa 9 luni atacurile cu aceleaşi simptome au recidivat şi au căpătat rapid un caracter serial. După numirea de tegretol la o doză de 400 mg pe zi, frecvența atacurilor a scăzut la 1 dată pe lună. Tegretol a fost înlocuit cu Depakine Chrono la o doză de 900 mg/zi, față de care s-a observat o remisiune clinică timp de 6 luni. Având în vedere simptomul clinic
VOLUM IV NUMĂRUL 3 2009
tomatice, limitarea crizelor la perioada de adormire, inteligența normală a pacientului, o reacție pozitivă la valproat, epilepsie occipitală idiopatică a fost diagnosticată.
La vârsta de 7 ani, crizele focale versive au reluat cu generalizare secundară la adormire cu aceeași frecvență de 1 dată pe lună. Creșterea dozei de Depakine la 1500 mg/zi nu a condus la o scădere a frecvenței convulsiilor. Când s-a adăugat lamiktal în doză de 75 mg/zi, atacurile au încetat timp de 4 luni, apoi s-au reluat cu aceeași frecvență. La vârsta de 8 ani s-au alăturat atacurile cu pierderea pe termen scurt a vederii. De la 8 ani 8 luni inainte de a adormi au inceput sa apara absente atipice: clipirea rapida cu inchiderea pleoapelor si instituirea globilor oculari in sus; conștiința fluctuează.
La vârsta de 9 ani au apărut multiple convulsii în serie, cu durată de câteva zile, cu halucinații vizuale simple sub formă de „rază” intermitentă în fața ochilor, cu ochii și capul întoarse spre dreapta. Înainte de a adormi, astfel de atacuri se transformau uneori în atacuri hemiclonice focale, care se manifestau prin reducerea feței.
musculatura pe dreapta, zvâcnirea capului spre dreapta, clonii ale membrelor drepte (mai mari decât brațul). Uneori, după atac, au existat o durere de cap puternică și vărsături. La aceeași vârstă au apărut convulsii inhibitorii: o aură sub formă de piele de găină la degetul mare al piciorului drept, urmată de slăbiciune de scurtă durată a piciorului drept și stângăcie a mâinii drepte. Topamax a fost introdus în regimul de tratament în doză de 100 mg/zi - nu au existat crize epileptice timp de 1 an.
De asemenea, la vârsta de 9 ani, au apărut pentru prima dată afecțiuni paroxistice, însoțite de dureri de cap severe, vărsături și dezvoltarea hemiparezei pe partea dreaptă. În unele cazuri, astfel de afecțiuni au fost însoțite de amauroză care a durat de la câteva minute la câteva zile.
La vârsta de 10,5 ani, atacurile au reapărut sub formă de întoarcere a capului la stânga, mișcări sacadate ale globilor oculari spre stânga, cu durata de până la 5 s, frecvență de până la 3 ori pe oră, zilnic, chiar și în timpul somnului. Doza de Topamax a fost crescută la 150 mg/zi fără efect semnificativ. La 10 ani 10 luni. dupa o durere de cap intensa, alternanta
Orez. 1. Pacient Ch.A. 10 ani. Diagnostic: sindrom MEAE. Epilepsie focală simptomatică.
Monitorizare video-EEG (2004): pe fondul unei încetiniri difuze a activității principale a creierului, a fost înregistrată activitate epileptiformă continuă în regiunea occipitală stângă. Modelele EEG subclinice ale unui atac au fost de asemenea înregistrate în regiunea occipitală stângă cu răspândire în regiunea temporală posterioară stângă.
Centrul de Neurologie și Epilepsie Pediatrică
sub îndrumarea profesorului K.Yu. Mukhina este implicată în diagnosticarea și tratamentul tulburărilor de anxietate ale sistemului nervos din Aetei, specializată în formele etiene de epilepsie.
Direcții principale
Activități:
Epilepsia la copii și adolescenți
Durere de cap
Tulburări de somn la copii
Tiki, enurezis
Examinarea copiilor în primele ^ luni de viață.
Examene în centrul nostru:
Diagnosticul și tratamentul bolilor sistemului nervos la copii
Diagnosticul complet (inclusiv prechirurgical) și tratamentul epilepsiei
Consultarea medicilor neurologi și epileptologilor
Consultarea unui medic pediatru (copii bolnavi frecvent, gastroenterologie etc.)
Consultarea unui psihiatru și psiholog.
Consultație genetică cu teste (inclusiv cariotipizare)
Monitorizare video-EEG (în sălile special echipate ale Centrului sau cu o vizită la domiciliul pacientului)
Electroencefalografie computerizată (digitală).
UZDG (dopplerografia cu ultrasunete) a vaselor capului și gâtului
Ecoencefalografie (ECHO EG)
Pe site-ul nostru vă puteți abona la revista „Russian Journal of Child Neurology” prin internet.
Informații detaliate despre activitatea Centrului de la 10:00 la 19:00 la telefon:
Tel.: (+7495) 983-09-03; (+7926)290-50-30 Tel./Fax: (+7495) 394-82-52
Adresa: str. Borisovskie Prudy, 13 ani, bld. 2. Internet: www.epileptologist.ru E-mail: [email protected](pentru o hartă detaliată a traseului, vezi site-ul web)
VOLUM IV NUMĂRUL 3 2009
convulsii hemclonice focale și secundare generalizate care au devenit în serie și au durat 48 de ore. Frizium a fost adăugat la topamax în doză de 10 mg/zi cu efect pozitiv temporar.
De la vârsta de 8 ani au început să se constate dificultăți în asimilarea materialului școlar; scăderea memoriei. Au fost crescute oboseala, epuizarea, inhibarea activității mentale. Băiatul a devenit capricios, iritabil, negativ; fundalul stării de spirit a scăzut. De la vârsta de 9 ani s-a înregistrat o creștere a acestei simptomatologie.
Din anamneza vieții, se știe că copilul s-a născut dintr-o a doua sarcină normală, un al doilea termen de naștere, greutatea la naștere 2800 g, lungime 53 cm. Dezvoltarea psihomotorie și a vorbirii timpurii a fost pe deplin adecvată vârstei. Boli anterioare: varicela la 6 ani, infectii virale respiratorii acute frecvente (de pana la 4 ori pe an) de la 6 ani. Ereditatea pentru epilepsie și alte boli neurologice nu este împovărată.
La momentul examinării (11 ani), starea copilului era gravă; reacţionează negativ la inspecţie. Conștient, pro-orientat
spatiu si timp. Intră în contact extrem de reticent, refuză să urmeze instrucțiunile. Nistagmus spontan la stânga, capul înclinat spre umărul stâng cu o întoarcere la dreapta. Limba este pe linia mediană, reflexul faringian este redus; Se notează disfagie și disartrie. Vederea este redusă.
Se determină hipotonie musculară difuză moderată. Reflexele tendinoase sunt reduse uniform. A existat o scădere ușoară a forței musculare la nivelul membrelor drepte. Reflexele patologice ale picioarelor nu au fost detectate. Nu există date obiective pentru încălcarea sensibilității. Nu merită în testul Romberg. Refuză să meargă. Când încerci să-l pui pe picioare, plânge, se așează pe podea. Lipsește la efectuarea unui test de index al degetelor. Vorbește încet, cu cuvinte simple, fără tragere de inimă.
Metode suplimentare de examinare. Monitorizare video-EEG (2004). Încetinirea semnificativă a activității principale de înregistrare în fundal. În timpul studiului, a fost înregistrată activitate epileptiformă continuă în regiunea occipitală stângă cu răspândire în regiunea temporală posterioară stângă și cu formarea periodică a unui model EEG.
născut în 1993 16/12/05
Orez. 2. Pacient Ch.A. 11 ani. Diagnostic: sindrom MELAS. Epilepsie focală simptomatică.
Monitorizarea video-EEG a fost efectuată în dinamică după 1 an (2005): o încetinire semnificativă a activității de fundal a creierului. În timpul înregistrării somnului, decelerația regională continuă este înregistrată în regiunea fronto-centrală dreaptă, în structura căreia se detectează activitatea de vârf a undelor în regiunea fronto-centrală dreaptă.
ARTICOLE ORIGINALE
stupa (Fig. 1). De asemenea, se determină decelerația regională continuă în regiunea fronto-centrală dreaptă cu includerea unor unde ascuțite unice.
Monitorizare video-EEG în dinamică (2005): încetinirea semnificativă a activității de fundal a creierului. Studiul a înregistrat o încetinire regională continuă în regiunea fronto-centrală dreaptă. În structura decelerației regionale în zona fronto-centrală dreaptă se evidențiază activitatea de vârf a undelor (Fig. 2).
RMN al creierului. Primul RMN (6 ani) a relevat un singur semnal hiperintens în modul T2 în emisfera stângă a cerebelului. Studiu RMN în timp (10,5 ani): a fost evidențiată o deteriorare semnificativă a leziunii primare odată cu răspândirea procesului patologic pe scară largă în regiunile occipital-parietale stângi și drepte ale ambelor emisfere ale creierului (profesor A.A. Alikhanov).
Potențiale evocate vizuale: modificări morfologice și funcționale semnificative ale sistemului aferent vizual la nivelul nervului optic și al părții corticale a analizorului vizual, mai pronunțate în stânga.
Consultație oftalmolog: atrofie parțială a nervilor optici. Elemente ale agnoziei corticale.
Electrocardiograma: ritm ectopic cu accelerare de până la 100 de bătăi pe minut.
Poziția verticală a axei electrice a inimii. Modificări ale proceselor de repolarizare, care sunt mai pronunțate în ortostazie.
Electroneuromiografia: a evidențiat tipul muscular primar de leziune. Vitezele de conducere de-a lungul nervilor periferici nu sunt reduse.
Studiul nivelului de lactat din sânge: conținutul de lactat din sânge este de 3,0 mmol / l (norma este de până la 1,8).
Ținând cont de prezența crizelor epileptice emanate din regiunile occipitale ale cortexului cerebral rezistente la terapie, episoade asemănătoare unui accident vascular cerebral, perioade de amauroză, declin cognitiv, prezența unor semnale hiperintense în cerebel și în regiunile posterioare ale cortexului cerebral pe RMN , o creștere a nivelului de lactat din sânge, pacientul a avut un diagnostic de sindrom MELAS a fost sugerat. În timpul unui examen genetic, mutația A3243G în stare heteroplasmatică a fost găsită în celulele sanguine (diagnosticul a fost efectuat la Centrul de Cercetare de Stat din Moscova al Academiei Ruse de Științe Medicale), iar diagnosticul a fost verificat.
Observarea în urmărire a arătat o progresie rapidă a încălcărilor funcțiilor mentale superioare, dezvoltarea orbirii corticale, imobilitatea completă a pacientului, urmată de debutul decesului la vârsta de 12 ani și 10 luni. (după 7 ani de la debutul bolii).
Bibliografie
1. Nikolaeva E.A., Temin P.A. Boli mitocondriale însoțite de dezvoltarea neuropsihică afectată. Sindromul MELAS // Tulburări ereditare ale dezvoltării neuropsihice a copiilor. Un ghid pentru medici editat de Temin P.A. Kazantseva L.Z. - Medicină, 2001. - S. 96-107.
2. Nikolaeva E.A., Temin P.A., Nikanorova M.Yu., Klembovsky A.I., Sukhorukov V.S., Dorofeeva M.Yu., Korsunsky A.A. Tratamentul unui copil cu sindrom mitocondrial MELAS (encefalopatie mitocondrială, acidoză lactică, episoade asemănătoare accidentului vascular cerebral) // Buletinul Rus de Perinatologie și Pediatrie. - 1997. - Nr. 2. - S. 30-34.
3. Smirnova I.N., Kistenev B.A., Krotenkova M.V., Suslina ZA. Curs asemănător unui accident vascular cerebral al encefalomiopatiei mitocondriale (sindrom MELAS) // Atmosfera. Boli nervoase. - 2006. - Nr. 1. - S. 43-48.
4. Temin PA, Nikanorova M.Yu., Nikolaeva E.A. Sindromul MELAS (encefalomiopatie mitocondrială, acidoză lactică, episoade asemănătoare accidentului vascular cerebral): manifestări principale, criterii de diagnostic, opțiuni de tratament // Nevrol. revistă - 1998. - Nr. 2. - S. 43-48.
5. Ajmone-Marsan C., Ralston B. Criza epileptică, morfologia funcțională și semnificația diagnostică. - Springfield (IL): Charles C. Thomas, 1957. - P. 3-231.
6. Aldrich M.S., Vanderzant C.W., Alessi A.G., Abou-Khalil B., Sackellares J.C. Orbire corticală ictală cu pierdere permanentă a vederii // Epilepsie. - 1989. - V. 30. - P. 116-20.
7. Araki T., Suzuki J., Taniwaki Y., Ishido K., Kamikaseda K., Turuta Y., Yamada T. A case of MELAS presenting complex partial status epilepticus // Rinsho Shinkeigaku. - 2001. - V. 41(8). - P. 487-90.
VOLUM IV NUMĂRUL 3 2009
8. Canafoglia L., Franceschetti S., Antozzi C., Carrara F., Farina L., Granata T., Lamantea E., Savoiardo M., Uziel G., Villani F., Zeviani M., Avanzini G. Epileptic fenotipuri asociate tulburărilor mitocondriale // Neurologie. - 2001. - V. 56(10). - P. 1340-6.
9. Chih-Ming Lin, Peterus Thajeb. Acidul valproic agravează epilepsia din cauza MELAS la un pacient cu o mutație A3243G a ADN-ului mitocondrial // Metab Brain Dis. - 2007 - V. 22(1). - P. 105-109.
10. Chinnery P.F., Howell N., Lightowlers R.N. et al. Patologia moleculară a MELAS și MERRF. Relația dintre încărcarea mutației și fenotipurile clinice // Creierul. - 1997. - V.120. - P. 1713-1721.
11. Durand-Dubief F., Ryvlin P, Mauguiere F. Polimorfismul epilepsiei asociat cu mutația A3243G a ADN-ului mitocondrial (MELAS): motive pentru diagnosticul întârziat // Rev Neurol (Paris). - 2004. - V. 160(8-9). - P. 824-829.
12. Dvorkin G., Andermann F., Carpenter S. Migrenă clasică, epilepsie intratabilă și accidente vasculare cerebrale multiple: un sindrom legat de encefalopatia mitocondrială / În: Andermann F., Lugaresi E., editori. migrenă și epilepsie. - Boston: Butterworths, 1987. - P. 203-32.
13. Fujimoto S., Mizuno K., Shibata H., Kanayama M., Kobayashi M., Sugiyama N., Ban K., Ishikawa T., Itoh T., Togari H., Wada Y. Serial electroencephalographs findings in patients cu MELAS // Pediatr Neurol. - 1999. - V. 20(1). - P. 43-48.
14. Goto Y., Nonaka I., Horai S.A. O mutație a genei tRNA leu(UUR) asociată cu subgrupul MELAS de encefalomiopatii mitocondriale // Nature. - 1990. - V. 348. - P. 651-653.
15. Hasuo K., Tamura S., Yasumori K., Uchino A., Goda S., Ishimoto S., et al. Tomografie computerizată și angiografie în MELAS (miopatie mitocondrială, encefalopatie, acidoză lactică și episoade asemănătoare AVC): raport de 3 cazuri // Neuroradiologie. - 1987.-V. 29. - P. 393-397.
16. Hirano M., Pavlakis S.G. Miopatie mitocondrială, encefalopatie, acidoză lactică și episoade de tip AVC (MELAS): Concepte actuale // J. clin. Neurol. - 1994. - V. 9. - P. 4-13.
17. Hori A., Yoshioka A., Kataoka S., Furui K., Tsukada K., Kosoegawa H., Sugianto, Hirose G. Crize epileptice la un pacient cu miopatie mitocondrială, encefalopatie, acid lactic și episoade asemănătoare accidentului vascular cerebral ( MELAS) // Jpn J Psihiatrie Neurol. - 1989. - V. 43(3). - P. 536-537.
18. Kuriyama M., Umezaki H., Fukuda Y., Osame M., Koike K., Tateishi J., et al. Encefalomiopatie mitocondrială cu creșterea lactat-piruvat și infarcte cerebrale // Neurologie. - 1984. - V. 34. - P. 72-77.
19. Kuzniecky R. Epilepsie simptomatică a lobului occipital // Epilepsie. - 1998. - V. 39 Suppl 4. - P. 24-31.
20. Ludwig B.I., Ajmone-Marsan C., Van Buren J. Depth and direct cortical recording in seizure disorders of extratemporal origin // Neurology. - 1976. - V. 26. - P. 1085-1099.
21. Ludwig B.I., Ajmone-Marsan C. Clinical ictal patterns in epileptic patients with occipital electroencephalo-graphic focuses // Neurology. - 1975. - V. 25. - P. 463-471.
22. Matthews P.M., Tampieri D., Berkovic S.F., Andermann F., Silver K., Chityat D., et al. Imagistica prin rezonanță magnetică arată anomalii specifice în sindromul MELAS // Neurologie. - 1991. - V. 41. - P. 1043-1046.
23. Miyazaki M., Saijo T., Mori K., Tayama M., Naito E., Hashimoto T., Kuroda Y., Nonaka I. Un caz cu MELAS asociat cu epilepsia partialis continua // No To Hattatsu. - 1991. - V. 23(1). - P. 65-70.
24. Montagna P., Gallassi R., Medori R., Govoni E., Zeviani M., Di Mauro S., et al. Sindromul MELAS: caracteristici migrenoase și epileptice și transmitere maternă // Neurologie. - 1988. - V. 38. - P. 751-754.
25. Ooiwa Y., Uematsu Y., Terada T., Nakai K., Itakura T., Komai N., et al. Fluxul sanguin cerebral în miopatia mitocondrială, encefalopatie, acidoză lactică și episoade asemănătoare accidentului vascular cerebral // AVC. - 1993. - V. 24. - P. 304-309.
26. Pavlakis S.G., Phillips P.C., Di Mauro S. et al. Miopatie mitocondrială, encefalopatie, acidoză lactică și episoade asemănătoare accidentului vascular cerebral: un sindrom clinic distinctiv // Un neurol. - 1984. - V. 16. - P. 481-488.
27. Ribacoba R., Salas-Puig J., Gonzalez C., Astudillo A. Characteristics of status epilepticus in MELAS. Analiza a patru cazuri // Neurologia. - 2006. - V. 21(1). - P. 1-11.
28. Williamson P.D., Spencer S.S. Caracteristici clinice și EEG ale crizelor parțiale complexe de origine extratemporală // Epilepsie. - 1986. - V. 27 (Suppl 2). - P. 46-63.
29. Williamson P.D., Thadani V.M., Darcey T.M., Spencer D.D., Spencer S.S., Mattson R.H. Epilepsia lobului occipital: caracteristici clinice, modele de răspândire a convulsiilor și rezultatele intervenției chirurgicale // Ann Neurol. - 1992. - V. 31. - P. 3-13.
30. Yi-Min Chen, Chih-Ming Lin, Peterus Thajeb. Efectul paradoxal al valproatului de sodiu care agravează epilepsia MELAS la un pacient cu mutația A3243G a ADN-ului mitocondrial // Central European Journal of Medicine. - 2007. - V. 2(1). - P.103-107.
31. Yoneda M., Maeda M., Kimura H., Fujii A., Katayama K., Kuriyama M. Vasogenic edem on MELAS: a serial study with diffusion-weighted MR imaging // Neurology. - 1999. - V. 53. - P. 2182-2184.
Sindromul MELAS este o tulburare mitocondrială caracterizată prin afectarea mușchilor și a SNC.
MELAS (English Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes - „mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes”) este o boală neurodegenerativă progresivă caracterizată prin manifestările enumerate în titlu și este însoțită de simptome polimorfe - stroke. , diabet, convulsii, scăderea pierderii auzului, boli de inimă, statură mică, endocrinopatii, intoleranță la efort și tulburări neuropsihiatrice.
Poveste.
Sindromul MELAS a fost descris pentru prima dată în 1984 de către Pavlakis și colegii; zece ani mai târziu, Pavlakis și Mizio Hirano au publicat o analiză a 110 cazuri.
tip de moștenire:
maternă
Epidemiologie:
Frecvența exactă a bolii nu este cunoscută. Există puține date în literatura de specialitate cu privire la incidența bolii. În nordul Finlandei, rata mutației A3243G este de 16,3:100.000.
Patogeneza:
Mutațiile ADN mitocondral, care controlează lanțul respirator al mitocondriilor, sunt însoțite de perturbarea fosforilării oxidative, cea mai importantă sursă de energie pentru procesele metabolice din celulă.
Manifestari clinice
La vârsta de 40 de ani, pacienții cu MELAS sunt internați cu o clinică de atac ischemic tranzitoriu, precum și cu epilepsie, vărsături repetate, cefalee și slăbiciune musculară. Acești pacienți sunt adesea diagnosticați clinic cu demență.
Vârsta fragedă și absența factorilor de risc specifici accidentului vascular cerebral fac MELAS mai atent.
Date de laborator
Acidoza lactată - niveluri crescute de lactat și piruvat.
Date de vizualizare
Modificările la nivelul creierului sunt similare cu modificările unui accident vascular cerebral.
Diferențele față de un accident vascular cerebral
1) zonele afectate nu coincid cu limitele bazinelor vasculare arteriale.
2) cu atacuri repetate, focarele sunt vizualizate într-o localizare diferită.
+ date clinice (varsta tanara, fara factori de risc pentru AVC).
CT
Zone hipodense multiple inconsistente cu patul vascular.
Calcificarea ganglionilor bazali (cel mai frecvent la pacienții în vârstă).
Atrofia apare pe fondul regresiei și îmbunătățirii clinice.
RMN
Infarct acut
Pentru diferențierea cu AVC se folosesc ADC și DWI (restricția de difuzie în AVC (edem citotoxic), iar în MELAS difuzia este ușor limitată sau nemodificată (edem vasogen).
Implicarea în procesul patologic al substanței albe subcorticale a creierului.
Deteriorarea în vizualizarea clarității contururilor circumvoluțiilor și creșterea semnalului de la acestea pe imaginile ponderate T2.
Infarct cronic
Modificările pot fi simetrice sau asimetrice.
Atrofia focală apare pe fondul regresiei și îmbunătățirii clinice.
Lobii parietal, occipital și temporal ai creierului sunt cel mai frecvent afectați.
spectroscopie MR
Niveluri crescute de lactat.



Materialele sunt destinate neurologilor, terapeuților și medicilor generaliști.
Serghei Likhachev, șef, MD. stiinte, profesor;
Inessa Pleshko, cercetător principal, Ph.D. Științe, Departamentul Neurologic al Centrului Republican Științific și Practic de Neurologie și Neurochirurgie.
Arteriopatia cerebrală autozomal dominantă cu infarcte subcorticale și leucoencefalopatie (CADASIL) este o boală autozomal dominantă progresivă, ale cărei manifestări clinice includ accidente vasculare cerebrale ischemice subcorticale recurente, migrene, demență subcorticală și tulburări afective. Prevalența actuală - 1 caz
la 100.000 de locuitori.
Centrul Republican Științific și Practic de Neurologie și Neurochirurgie vede 7 pacienți (inclusiv 4 femei) cu CADASIL; vârsta - de la 32 la 68 de ani. Au fost examinați prin metode neurologice, genetice moleculare. Au existat simptome caracteristice; în istorie - migrene, accidente vasculare cerebrale lacunare recurente și tulburări afective. RMN cerebral a evidențiat infarcte subcorticale și leucoencefalopatie caracteristice CADASIL.
Ca rezultat al diagnosticului genetic molecular, 2 persoane au avut o mutație heterozigotă a genei Notch3 de pe cromozomul al 19-lea, care provoacă CADASIL. Genele Notch codifică receptorii transmembranari implicați în ontogeneza celulară. Cu CADASIL, în cele mai multe cazuri, sunt determinate mutații missense, din cauza cărora structura proteinei transmembranare se modifică și funcțiile acesteia sunt afectate.
Patogenia CADASIL nu este complet clară. Se crede că principalul factor este arteriopatia cu ocluzie progresivă a micilor vase perforante ale substanței albe a creierului (care duce la hipoperfuzie cronică). În același timp, se constată incluziuni osmiofile granulare caracteristice, determinând proliferarea componentelor membranei bazale, îngroșarea membranei medii și compresia mecanică a arterelor mici. Ca urmare, bariera hemato-encefalică este deteriorată - se dezvoltă edem.
Un factor patologic suplimentar este activarea astrocitelor în apropierea peretelui vascular. Ei eliberează endoteliul-1, provocând vasoconstricție și afectarea fluxului sanguin.
Compoziția incluziunilor osmiofile granulare este necunoscută. Se presupune că proteina Notch3 este una dintre componentele lor. În biopsiile de piele ale pacienților cu o mutație Notch3, granulele osmiofile și degenerarea celulelor musculare netede pot fi detectate chiar înainte de vârsta de 20 de ani.
Diagnosticul clinic al CADASIL:
- istoric familial împovărat;
- dezvoltarea primelor simptome ale bolii înainte de vârsta de 50 de ani;
- prezența a două dintre următoarele simptome - migrenă, accidente vasculare cerebrale recurente, tulburări de dispoziție, demență subcorticală.
Factorii de risc vascular asociati etiologic cu simptomele neurologice trebuie exclusi. RMN-ul arată afectarea substanței albe a emisferelor cerebrale și absența infarctelor corticale.
Un diagnostic de încredere al „CADASIL” este confirmat de un rezultat pozitiv al diagnosticului genetic molecular sau de detectarea arteriopatiei cu incluziuni osmiofile granulare caracteristice în biopsia cutanată sau musculară.
Cele mai frecvente simptome ale CADASIL sunt atacurile ischemice tranzitorii și accidentele vasculare cerebrale ischemice, observate la aproape 85% dintre pacienți.
Se caracterizează printr-o evoluție recurentă, manifestată prin sindroame clasice de accidente vasculare cerebrale lacunare și remisiune clinică completă după câteva zile sau săptămâni.
Al doilea cel mai frecvent sunt tulburările cognitive (observate la 60% dintre pacienți). Poate începe la vârsta de 35 de ani, uneori chiar înainte de episoadele ischemice. Aproximativ 75% dintre pacienții cu CADASIL dezvoltă demență. Primul simptom este de obicei o migrenă; apare adesea înainte de vârsta de 20 de ani și de obicei precede accidentul vascular cerebral.
Datele privind implicarea inimii în procesul patologic în CADASIL sunt contradictorii. L. Oberstein şi colab. (2003) au descoperit că 25% dintre pacienții diagnosticați cu CADASIL au avut antecedente de infarct miocardic acut sau patologie unde Q pe electrocardiogramă. Într-un alt studiu, Cumurciuc et al. (2006) nu au găsit niciun istoric cardiac pozitiv la 23 de persoane cu o mutație Notch3.
Manifestările clinice ale CADASIL și microangiopatia cerebrală de altă etiologie sunt similare - este necesar diagnosticul diferențial.
Pentru determinarea în timp util a CADASIL la pacienți și familiile acestora, este necesar să se recurgă la metode genetice moleculare și/sau studii histologice.
sindromul MELAS
Encefalomiopatia mitocondrială cu acidoză lactică și episoade asemănătoare accidentului vascular cerebral (MELAS) este o boală ereditară rară cauzată de patologia genomului mitocondrial, afectarea metabolismului energetic și funcționarea celor mai dependente de organe și țesuturi (SNC, mușchi cardiaci și scheletici, ochi, rinichi, ficat, măduvă osoasă, sistemul endocrin). Variabilitatea largă a manifestărilor clinice ale sindromului MELAS și apariția rară pretermină dificultăți de diagnosticare pentru medic.
În Centrul Republican Științific și Practic de Neurologie și Neurochirurgie, 3 pacienți (o femeie de 46 de ani și fiii ei, de 24 și 23 de ani) sunt observați cu sindrom MELAS diagnosticat. Au fost supuși unui examen clinic și neurologic, diagnostic genetic molecular, RMN al creierului.
Toate sunt scurte; în istorie - simptome ale patologiei mitocondriale: hipoacuzie neurosenzorială, cefalee asemănătoare migrenei, toleranță slabă la efort. Debutul bolii este convulsii convulsive generalizate. La 2 pacienţi, primele simptome au apărut înainte de vârsta de 20 de ani; au fost convulsii epileptice succesive, episoade de afectare vizuală cu prezența focarelor în neuroimagistică în regiunile occipitală și temporală, creșterea nivelului de lactat în sânge și lichid cefalorahidian. 1 persoană a avut o scădere moderată a funcțiilor cognitive; conform ecografiei inimii - cardiomiopatie hipertrofică; Diabet.
Un studiu genetic molecular a evidențiat leziuni multisistem tipice MELAS, variabilitate largă și severitate variabilă a manifestărilor clinice, corespunzătoare numărului de copii mutante A3243G din gena tRNA Leu(UUR).
MELAS se caracterizează printr-un tip de moștenire maternă, prezența unor cazuri sporadice când apare o mutație de novo; acumularea în celule - atât de tip normal, cât și de tip mutant - a ADN-ului mitocondrial (heteroplasmie) și distribuția aleatorie în timpul diviziunii între celulele fiice (segregare mitotică). La nivel genetic, cauza sindromului MELAS este rearanjarea heteroplasmatică 3243A>G în gena tRNALeu(UUR) (80% din cazuri sunt detectate).
Patogenia bolii nu a fost încă studiată. Există 2 teorii principale - „angiopatia mitocondrială” și „citopatia mitocondrială”. Se știe că leziunea asemănătoare accidentului vascular cerebral nu corespunde zonelor vasculare și se extinde în zonele învecinate din cauza edemului vasogenic concomitent cauzat de activitatea epileptică prelungită. După cum sa sugerat, episoadele asemănătoare unui accident vascular cerebral se datorează hiperexcitabilității neuronale într-o zonă limitată a creierului. Ea apare din disfuncția mitocondrială în celulele endoteliale capilare, sau în neuroni, sau în astrocite; depolarizează neuronii adiacenți, ducând la răspândirea activității epileptice.
În plus, în intervalele dintre episoadele de tip stroke-like, conform tomografiei computerizate cu emisie de foton unic (SPECT), la pacienții cu MELAS, se observă hipoperfuzie a cortexului cingulat posterior, ceea ce indică o tulburare a hemodinamicii cerebrale.
Încălcarea fosforilării oxidative, ruperea lanțului respirator mitocondrial contribuie la predominarea metabolismului catabolic și modificările de la ciclul Krebs la glicoza anaerobă cu acumulare de lactat. Un nivel ridicat al acestuia din urmă în SNC se corelează de obicei cu perioadele de simptome neurologice.
Principalele semne clinice ale MELAS sunt episoade asemănătoare accidentului vascular cerebral, acidoza lactică și prezența „fibrelor roșii rupte” în probele de biopsie musculară. Manifestări suplimentare pot fi demență, psihoză, convulsii epileptice, cefalee asemănătoare migrenei, ataxie, miopatie, calcificarea ganglionilor bazali la neuroimagistică, atrofie optică, retinopatie, surditate, diabet, pseudo-obstrucție intestinală, cardiomiopatie.
Vârsta timpurie a debutului MELAS este de la 5 la 20 de ani, cu toate acestea, există observații ale unui debut tardiv - în deceniile 5-6 de viață. Există cazuri când sindromul a început după tulburări cardiace.
Natura multisistemică a leziunilor din MELAS complică diagnosticul clinic.
Natura ereditară a bolii obligă să efectueze studii genetice moleculare pentru a pune un diagnostic precis.
și identificați alți pacienți - dintre rudele pacientului.
Materialele sunt destinate neurologilor, terapeuților și medicilor generaliști.
























1 din 24
Prezentare pe tema: sindromul MELAS
diapozitivul numărul 1

diapozitivul numărul 2

Descrierea diapozitivului:
Sindromul MELAS (ing. Encefalomiopatie mitocondrială, acidoză lactică și episoade asemănătoare AVC - „encefalomiopatie mitocondrială, acidoză lactică, episoade asemănătoare AVC”) este o boală neurodegenerativă progresivă caracterizată prin manifestările enumerate în titlu și este însoțită de simptome polimorfe - diabet, convulsii, pierderea auzului, boli de inimă, statură mică, endocrinopatii, intoleranță la efort și tulburări neuropsihiatrice.
diapozitivul numărul 3

Descrierea diapozitivului:
Istoric Sindromul MELAS a fost identificat pentru prima dată ca o formă independentă din punct de vedere nosologic de către S. Pavlakis et al. în 1984. Cu toate acestea, există motive să credem că boala a fost descrisă mai devreme sub denumirea de „poliodistrofie de familie, miopatie mitocondrială, acidemia lactică”. Până în 1994, 110 observații ale sindromului MELAS au fost publicate în literatură.
diapozitivul numărul 4

Descrierea diapozitivului:
diapozitivul numărul 5

Descrierea diapozitivului:
Etiologie, patogeneză MELAS se referă la bolile mitocondriale. Sindromul se dezvoltă ca urmare a mutațiilor punctuale ale ADN-ului mitocondrial. Localizarea mutațiilor în 3 puncte asociate cu sindromul MELAS a fost dezvăluită: două în ARN de transfer și una în citocrom c oxidază.
diapozitivul numărul 6

Descrierea diapozitivului:
diapozitivul numărul 7

Descrierea diapozitivului:
diapozitivul numărul 8

Descrierea diapozitivului:
Modificări patologice Un semn patologic caracteristic al sindromului MELAS, precum și o serie de alte encefalomiopatii mitocondriale (sindroame Kearns-Sayre, MERRF etc.), sunt fibrele roșii „rupte” (RRF), care apar în țesutul muscular cu o modificare modificată. Pată tricrom Gomory. Sunt substratul morfologic al leziunilor ADN mitocondrial și se formează ca urmare a proliferării mitocondriilor anormale. Fibrele roșii „rupte” sunt rezultatul mutațiilor care deteriorează genele ARN de transfer și duc la perturbarea sintezei proteinelor intramitocondriale. S-a demonstrat că astfel de caracteristici morfologice ale țesutului muscular precum prezența vaselor cu activitate ridicată a succinat dehidrogenazei și o cantitate semnificativă de fibre musculare pozitive pentru citocrom c-oxidază sunt o trăsătură caracteristică a sindromului MELAS, care fac posibilă diferențierea. este din sindroamele Kearns-Sayre și MERRF. Unul dintre cele mai specifice semne de afectare a creierului în această boală este prezența focarelor vechi și noi de infarcte.
diapozitivul numărul 9

Descrierea diapozitivului:
fibre roșii „rupte” (RRF)
diapozitivul numărul 10

Descrierea diapozitivului:
Tabloul clinic Primele semne apar adesea la vârsta de 6-10 ani, deși sunt posibile atât un debut mai devreme al bolii (până la 2 ani), cât și unul mai târziu (21-40 de ani). Înainte de apariția primelor semne ale bolii, majoritatea pacienților se dezvoltă normal. Manifestări clinice inițiale: convulsii, dureri de cap recurente, vărsături, anorexie, intoleranță la efort, tulburări psihice, simptome neurologice (pareză, ataxie etc.).
diapozitivul numărul 11

Descrierea diapozitivului:
Tabloul clinic Intoleranța la activitatea fizică, după care starea de sănătate se înrăutățește, apare slăbiciune musculară, uneori mialgie. Episoadele asemănătoare accidentului vascular cerebral se manifestă prin atacuri recurente de cefalee, amețeli, dezvoltarea simptomelor neurologice focale (pareză, paralizia extremităților, nervii cranieni) și comă. Convulsiile din sindromul MELAS sunt foarte variabile – paroxisme focale, tonico-clonice generalizate, mioclonie. Convulsiile sunt puțin sensibile la terapia anticonvulsivante.
diapozitivul numărul 12

Descrierea diapozitivului:
Tabloul clinic Demența se dezvoltă de obicei odată cu progresia bolii, dar relativ rar acţionează ca un simptom manifest. Complex de simptome miopatice (slăbiciune musculară, oboseală, uneori malnutriție). Odată cu debutul timpuriu al bolii, cursul acesteia este mai malign. Deci, odată cu debutul MELAS înainte de vârsta de 20 de ani, rata mortalității este de 30%.
diapozitivul numărul 13

Descrierea diapozitivului:
Principalele criterii de diagnostic sunt: intoleranța la efort; debutul bolii înainte de vârsta de 40 de ani (de obicei înainte de vârsta de 20 de ani); episoade asemănătoare accidentului vascular cerebral; convulsii; fibre roșii „rupte” în biopsiile mușchilor scheletici; acidoză lactică; demență progresivă; sindrom miopatic; statură mică; surditate.
diapozitivul numărul 14

Descrierea diapozitivului:
Criterii suplimentare de diagnostic: calcificarea ganglionilor bazali pe tomografia computerizată (CT) sau imagistica prin rezonanță magnetică (RMN) a creierului; ataxie; comă; atrofie a nervului optic; retinită pigmentară; sindrom Wolff-Parkinson-White; insuficiență cardiacă; oftalmoplegie externă progresivă ; tulburări de conducere ale inimii; diabet zaharat.
diapozitivul numărul 15

Descrierea diapozitivului:
Date ale studiilor de laborator și funcționale: Un semn caracteristic al bolii este detectarea acidozei lactice în sânge și lichidul cefalorahidian. La jumătate dintre pacienții din lichidul cefalorahidian este detectată o creștere a nivelului de lactat și proteine.Este important studiul enzimelor lanțului respirator, mai des sunt depistate modificări ale activității enzimelor complexului I. ECG: conducere cardiacă tulburări, poate fi detectat sindromul Wolff-Parkinson-White. CT a creierului: zone de infarct mai des în emisferă, mai rar în cerebel, ganglioni bazali. Poate exista calcificare a ganglionilor bazali, atrofie a cortexului cerebral. Angiografia cerebrală: o creștere a calibrului vaselor de sânge (artere, vene, capilare).
diapozitivul numărul 16

Descrierea diapozitivului:
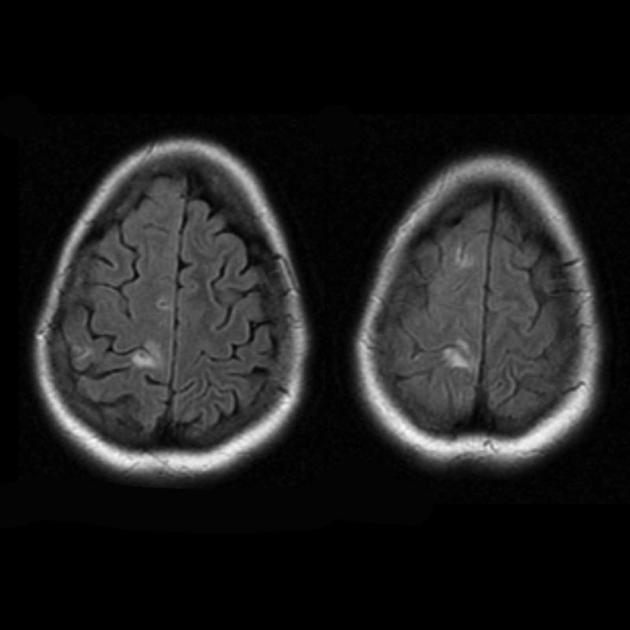
RMN cerebral al pacientului A., imagini ponderate T2. a, b – primul studiu: focare simetrice de intensitate crescută a semnalului în proiecția lobilor parietali ai ambelor emisfere. c, d – al 2-lea studiu: în proiecția lobilor temporali și parietali ai emisferei drepte se constată o extindere a zonei semnalului RM alterat. În emisfera stângă în proiecția lobului parietal, dimensiunea focarului patologic a scăzut semnificativ.
diapozitivul numărul 17

Descrierea diapozitivului:
CT a creierului: în lobul temporal al emisferei stângi cu extensie parțială până la lobul parietal se determină un focar de densitate ușor redusă (săgeată). În ambele emisfere se determină focare de densitate crescută (calcificări) în regiunea nucleilor lenticulari și a tuberculilor vizuali.
diapozitivul numărul 18

Descrierea diapozitivului:
Studiul perfuziei cerebrale prin SPECT la pacientul A. a-c - studiu 11/12/2003 - zona de perfuzie scăzută (albastru și verde) în regiunea temporală a emisferei drepte (a) și perfuzie mare (roșu) în lobii occipitali ( b) și lobul temporal al emisferei stângi (c). d–f – studiu din 18 februarie 2004 – scăderea perfuziei în lobul temporal stâng (f) comparativ cu studiul anterior.
Descrierea diapozitivului:
Tratament simptomatic Pentru corectarea defectelor biochimice, coenzima Q10 (80 - 300 mg/zi), vitaminele K1, K3 - filochinona (25 mg/zi) si menadiona (pana la 75 mg/zi), acid succinic (pana la 6). mg/zi) se folosesc vitamina C (2-4 g/zi) si alte vitamine (riboflavina, tiamina, nicotinamida). Se știe că coenzima Q10 în condiții fiziologice transferă electroni de la complexele I și II la complexul III și contribuie astfel la stabilizarea lanțului respirator, la scăderea nivelului de lactat și piruvat. Vitaminele K1 și K3 sunt în mod evident capabile să îndeplinească funcția de transport de electroni la nivelul complexelor I și III. Acidul succinic asigură transferul de electroni ai complexului II. Vitamina C este considerată un donator de electroni al complexului IV, precum și un antioxidant important. În plus față de acidul ascorbic, vitamina E (300 - 500 mg / zi) este prescrisă pentru a preveni deteriorarea radicală a oxigenului a membranelor mitocondriale.
diapozitivul numărul 21

Descrierea diapozitivului:
Tratament Pentru a stimula sinteza ATP, se propune utilizarea idebenonei (90 - 180 mg/zi), care are proprietatea de a intensifica metabolismul energetic in tesutul creierului. Introducerea vitaminelor riboflavină (100 mg / zi) și nicotinamidă (până la 1 g / zi) - precursorii coenzimelor NAD și FAD, care sunt implicate activ în procesele oxidative, ajută, de asemenea, la îmbunătățirea producției de energie a mitocondriilor. Din cauza deficitului secundar de carnitină, pacienților li se prescrie L-carnitină (până la 100 mg/zi). Pentru a reduce nivelul de lactat din sange si lichidul cefalorahidian se foloseste dicloracetat de sodiu (25-100 mcg/kg) Folosind teste de laborator, este necesar sa se verifice eventualele disfunctii ale sistemului endocrin (diabet zaharat, hipoparatiroidism) și sistemul cardiovascular (blocadă). Dacă sunt detectate încălcări, se efectuează corectarea medicală a acestora.
diapozitivul numărul 24

Descrierea diapozitivului:
Buletinul AGIUV, număr special, 2013 udk 616.8-007: 616.853.3
debutul sydroml melas cu convulsii febrile
(studiu de caz)
Acea. Musabekova, A. i. Khamzina
Universitatea Slavă Kârgâz-Rusă, Departamentul de Neurologie și Neurochirurgie, Bishkek, Kârgâzstan
Convulsiile febrile (FS) sunt cunoscute încă din antichitate. Hipocrate a mai scris că FS apare cel mai adesea la copiii din primii 7 ani de viață și mult mai rar la copiii mai mari și la adulți. Dar pentru prima dată termenul „convulsii febrile” a fost folosit în 1904 de B. Hochsinge pentru a se referi la paroxismele convulsive care se dezvoltă în copilărie pe un fundal de febră. În prezent, este de preferat să vorbim despre convulsii febrile (FA) mai degrabă decât despre FS, deoarece în tabloul clinic al acestei afecțiuni se pot observa nu numai paroxisme convulsive, ci și non-convulsive 2]. Conform definiției ILAE din 1993, FA este o convulsie care apare la copiii cu vârsta peste 1 lună asociată cu o boală febrilă care nu este cauzată de infecția SNC; fără convulsii neonatale anterioare și convulsii neprovocate și neîndeplinirea criteriilor pentru alte convulsii simptomatice acute. Conform proiectului de clasificare din 2001, FA este clasificată ca un grup de afecțiuni care nu necesită un diagnostic obligatoriu de epilepsie. Astfel, FA este definită ca un episod de crize epileptice care apare la copiii cu vârsta de 6 luni și peste. până la 5 ani cu creșterea temperaturii în timpul unei boli virale sau bacteriene care nu este asociată cu neuroinfecție și tulburări metabolice. Adevărata FA trebuie distinsă de convulsiile induse de febră, care pot face parte din structura unui număr de forme de epilepsie, cum ar fi sindromul Dravet. În cazuri rare, FA poate fi primul simptom al bolii mitocondriale la copii.
Sindromul MELAS (encefalomiopatie mitocondrială cu acidoză lactică și episoade asemănătoare accidentului vascular cerebral) a fost identificat pentru prima dată ca formă nosologică separată de S. Pavlakis și colab. abia în 1984. Boala aparține grupului de boli mitocondriale asociate cu mutații punctuale ale ADN-ului mitocondrial, care duc la o încălcare a producției de energie în lanțul respirator mitocondrial. Se știe că mutațiile punctuale pot apărea în multe gene (MTTL1, MTTQ, MTTH, MTTK, MTTS1, MTND1, MTND5, MTND6, MTTS2) și pot fi moștenite prin linia maternă. Prevalența sindromului MELAS este greu de estimat din cauza varietății de manifestări și a dificultății asociate în diagnostic. până în 2000, au fost publicate peste 120 de observații ale bolii. Simptomele cardinale ale sindromului MELAS sunt: intoleranța la efort, episoade asemănătoare unui accident vascular cerebral, convulsii, fibre „roșii rupte” în biopsiile țesutului muscular, acidoza lactică și debutul bolii înainte de vârsta de 40 de ani. Sindromul MELAS trebuie diferențiat de alte boli mitocondriale: sindromul Kearns-Seyer și MERRF.
Mai jos este propria noastră observație a pacientului P., născut în 2003, care locuiește în Bishkek. Un copil a venit la noi în centrul clinicii MEBI LTD din Bishkek în primăvara anului 2013 cu plângeri de convulsii tonico-clonice la picioare și brațe care durează până la 2 minute, care apar cu pierderea conștienței și în curs de dezvoltare.
numai pe fondul creșterii temperaturii corpului peste 37 ° C, precum și a apariției dificultății în stăpânirea materialului școlar, pierderi de memorie, oboseală crescută și slăbiciune musculară, stângăciune la mers.
Debutul bolii la o fată a fost observat la vârsta de 6 luni cu o criză tonico-clonică generalizată care durează până la 1 minut pe fondul unei creșteri a temperaturii corpului la 38 ° C, după care a fost internată în Republica Populară. Spitalul de Boli Infecțioase din Bishkek, unde a fost exclusă o neuroinfecție. Ulterior, AF a apărut de fiecare dată când temperatura corpului a crescut peste 37°C. La vârsta de 1 an, la contactarea Centrului Național de Pediatrie și Chirurgie Pediatrică al Republicii Kârgâz, s-a efectuat un RMN al creierului, EEG, unde nu s-a depistat nicio patologie, s-a prescris depakine în doză de 20 mg/kg/ zi. Cu toate acestea, FA a continuat în timp ce luați un medicament anticonvulsivant. La vârsta de 5 ani, au aplicat independent la Spitalul Clinic Republican de Copii (RCCH) din Moscova, unde au fost repetate RMN-ul creierului și monitorizarea video-EEG (VEM) a somnului în timpul zilei, unde din nou nu a fost detectată nicio patologie. Medicii RCCH l-au diagnosticat cu epilepsie criptogenă și i s-a recomandat creșterea dozei de depakine la 25 mg/kg/zi. Primele trei clase le-am terminat într-o școală cuprinzătoare cu clasele „4” și „5”. Începând cu vârsta de 9 ani, mama a început să observe o creștere treptată a oboselii rapide a copilului după efort fizic, apariția dificultății în însușirea materialului școlar, s-a pus problema transferului copilului într-o instituție școlară specializată pentru copiii cu retard mintal. FA a continuat să deranjeze copilul chiar și după 7 ani în timp ce lua depakin chrono în doză de 25 mg/kg/zi.
Din anamneza vieții: un copil din prima sarcină care a procedat pe fondul toxicozei ușoare în primul trimestru, a avut un episod de SARS fără febră într-o perioadă de 5 luni. Naștere la termen, independent în prezentație cefalică, scor Apgar 7/8 puncte, CM - 3340 gr., înălțime - 52 cm.Dezvoltarea timpurie a copilului a corespuns normei de vârstă.
La momentul examinării la clinică la vârsta de 10 ani, din partea nervilor cranieni, a existat o ușoară deviație a limbii spre dreapta, sindrom miopatic la nivelul brațelor și picioarelor sub formă de hipotensiune arterială, hipotrofie ușoară a brațele și picioarele proximale cu o scădere a forței musculare de până la 4 puncte, o scădere a reflexelor tendinoase, precum și o ușoară eșalonare în poziția Romberg și stângaci la efectuarea testelor deget-nas și genunchi-călcâi, o scădere a memorie de termen și atenție.
Examene suplimentare: Hb 112 g/l, eritrocite 3,5*10”12/l, analize hepatice, proteine totale, zahar, creatinina in limite normale.
Continuarea FA după 5 ani cu dezvoltarea rezistenței la valproat, adăugarea unui sindrom miopatic și scăderea funcțiilor cognitive au făcut posibilă sugerarea că pacientul ar putea avea patologie mitocondrială și anume sindromul MELAS, care ar necesita
pentru a efectua o serie de studii suplimentare. Electroneuromiografia efectuată în NCP și DC a evidențiat un tip de leziune musculară primară sub forma unei scăderi a duratei potențialului unităților motorii cu 30-35% și o scădere a amplitudinii acestora cu o viteză normală de conducere de-a lungul nervilor periferici. . Cu VEM repetat, nu a fost detectată nicio patologie. În laboratorul SVS numit după V.M. Orașul Savinov din Almaty, concentrația de depakine în sânge înainte de a lua medicamentul a fost determinată - 86,98 ng / ml și la 2 ore după administrarea medicamentului - 113,61 ng / ml la o rată de 50-100 ng / ml. În clinica Privat din Almaty, conținutul de acid lactic pe stomacul gol a fost determinat - 3,1 mmol/l, cu o rată de până la 1,7 mmol/l. S-a pus un diagnostic preliminar de sindrom MELAS, s-a înlocuit depakina cu keppra, s-a introdus coenzima Q10, carnitină, vitaminele B, vitamina E, s-a recomandat o dietă restrictivă în carbohidrați și s-a recomandat un examen genetic.
Astfel, în cazul clinic prezentat de noi, copilul avea FA simplă, pentru tratamentul profilactic căruia i s-a prescris depakina cu utilizare pe termen lung timp de câțiva ani în ciuda prezenței rezistenței la medicamente. Ținând cont de lipsa unor dovezi clare privind eficacitatea utilizării profilactice a anticonvulsivantelor la copiii cu FA, numirea depakinei în acest caz a fost inadecvată. Deci, conform literaturii de specialitate, utilizarea pe termen lung a depakinei și barbituricelor agravează semnificativ evoluția bolilor mitocondriale, ducând uneori la progresia procesului patologic, ceea ce s-a întâmplat în cazul nostru clinic.
bibliografie:
1. Guzeva V.I. Sindroame speciale (crize situaţionale) / V.I. Guzeva // Epilepsie și stări paroxistice non-epileptice la copii.-M.: MIA, 2007.- P. 443-457
2. Mukhin K.Yu. Convulsii febrile / A.S. Petrukhin
// Neurologia copilăriei. - M.: Medicină, 2004.-S.664-668.
3. Nikanorova M.Yu., Temin P.A., Kobrinsky B.A. Convulsii febrile / P.A. Temina, M.Yu. Nikanorova // Epilepsie și sindroame convulsive la copii.- M.: Medicină, 1999.- P. 169-195.
4. Principalele metode de tratament ale copiilor care suferă de boli mitocondriale: Metodică. instrucţiuni.-M., 2001.
5. Temin P.A. etc. //Nevrol. jurnal - 1998. Nr. 2. - P. 43.
6. Yakhno N.N. etc. //Nevrol. revistă 1998.- Nr 5.- P. 14.
7 Ban S. et al. // Act Pathol. Jpn.- 1992.-Vol. 42.-p. 818.
8. Hirano M., Pavlakis S.G. // J. Clin. Neurol. -1994.-Vol. 9.-p. 4.
9. Raportul Comisiei ILAE: glosar de terminologie descriptivă pentru semiologia ictală: raport al Grupului operativ ILAE privind clasificarea și terminologia / Epilepsia.- 2001.- Vol. 42.-P.1212-1218.
10.ILAE. Ghid pentru studii epidemiologice asupra epilepsiei /Epilepsie.- 1993.- Vol.34.- P. 592-596.
11. Pavlakis S.G. et al. //Ann. Neurol.-1984. -Vol. 16.-P. 481.
12. Sciacco M. et al. // J. Neurol. -2001. -V 248. -P. 778.
Crizele febrile pot fi adesea primul simptom al bolilor mitocondriale la copii, ceea ce complică foarte mult diagnosticul și inițierea în timp util a tratamentului patogenetic al bolii și, uneori, provoacă utilizarea de medicamente care agravează cursul și prognosticul bolii.
Cuvinte cheie: convulsii, terapie.
Crizele febrile pot fi adesea primul simptom al bolii mitocondriale la copii, ceea ce complică foarte mult diagnosticul în timp util și tratamentul precoce și, uneori, determină utilizarea medicamentelor să agraveze cursul și prognosticul bolii.
Cuvinte cheie: convulsii, terapie.
udk 616.831-005.4
CAZURI DE AVC ISCHEMIC CA MANIFESTARE A ENCEFALOPATIEI MITOCONDRIALE LA UN PACIEN TANAR
VÂRSTĂ
Karbozova K.Z., Lutsenko I.L.
Departamentul de Neurologie cu un curs de genetică medicală, Academia Medicală de Stat Kârgâză, numită după I.K. Akhunbaeva,
Bishkek, Kârgâzstan
Prevalența accidentului vascular cerebral la o vârstă fragedă (până la 45 de ani) este de la 2,5 până la 10% din toate cazurile de accidente cerebrovasculare și continuă să crească. La pacientii tineri, cele mai frecvente cauze ale tulburarilor vasculare ischemice sunt: anomalii ale sistemului cerebrovascular, disectia, patologia cardiaca, migrena, defecte de coagulare, AFLS,.
În ultimele 5 luni, 608 de pacienți au fost tratați în Departamentul de Neurologie nr. 1 al Spitalului Național din subordinea Ministerului Sănătății al Republicii Kârgâzie (NHMZKR). S-a făcut o analiză a 46 (7,5) anamneză de pacienți cu AVC ischemic, dintre care 4 (8,7%) erau tineri (sub 45 de ani conform OMS). Tabelul 1 enumeră subtipurile de accident vascular cerebral ischemic.
Prezentăm istoricul unui pacient internat cu un diagnostic inițial de accident cerebrovascular acut (ACI), la care adevărata natură a bolii a putut fi stabilită doar în timpul observației dinamice și a unei examinări suplimentare speciale.
Tabelul 1.
Subtip de accident vascular cerebral Număr de pacienți în %
Aterotrombotic 32 69,5
Lacunar 6 13.04
Hemoreologice 3 6.5
Cardioembolic 3 6.5
Mitocondriale 1 2.2